Il n'est pas possible de séquencer un génome en un seul passage. La molécule d'ADN est beaucoup plus grande que les capacités des séquenceurs. Il faut donc couper le génome en fragments et séquencer chacun de ces fragments. Vous demanderez à votre enseignant de biologie moléculaire comment en pratique celà se fait. Ici on supposera que la pratique expériementale conduit à la fabrication de fragments tous de même longueur L et dont l'extrémité droite (ou gauche au choix) est un tirage "au hasard" d'un point dans le génome. Bien entendu votre enseignant de biologie vous dira, horrifié, "Mais c'est beaucoup plus compliqué que celà!!!". Vous répondrez que vous ferez un peu mieux quand vous attaquerez la deuxième fiche sur le sujet.
Les fragments peuvent se recouvrir partiellement pour définir des zones de séquences ainsi connues appelées "contig". L'objectif final est que le génome lui même ne constitue qu'un seul contig.
Ainsi on dispose de:
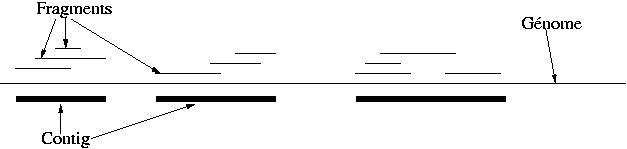
Le taux de couverture est la longueur totale séquencée raportée à la taille du génome. Bien entendu on ne peut espérer couvrir l'ensemble du génome que si A est plus grand que 1.
Ce que l'on veut:
Dans cette première partie vous avez besoin de connaître,
Dans toute la suite on suppose que G est très grand par rapport à L. Ceci permettra d'ignorer ce qui se passe aux extrémités du génome (un fragment ne peut pas commencer avant le début du génome ou finir après la fin!).
Les extrémités gauches des fragments se répartissent uniformément le long du génome. La probabilité que l'extrémité gauche d'un fragment appartienne au segment [x-h,x] est égale au rapport de la longueur de ce segment divisé par la longueur totale du génome, soit h/G.
Le nombre de fragments commençant entre la position x-h et la position x suit donc la loi binomiale B(N,h/G).
N est grand et si h est supposé petit devant G cette binomiale est très voisine d'une loi de Poisson P(Nh/G) que nous allons utiliser pour les calculs suivants.
Si on choisi h=L, le nombre de fragments de longueur L dont l'extrémité gauche appartient à [x-L,x] est exactement le nombre de fragments recouvrant la position x du génome.
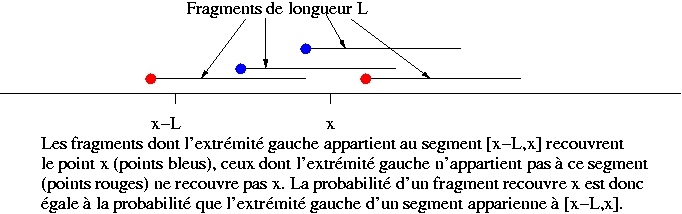
La probabilité qu'une position du génome ne soit pas recouverte est donc exp(-NL/G) = exp(-A)
On en déduit un des résultats recherchés: la probabilité qu'une position soit déterminée lors d'un séquençage avec un taux de recouvrement A est 1-exp(-A)
Un autre résultat peut être obtenu avec des raisonnements mathématiques de même nature: le nombre moyen de contigs. En remarquant que le nombre de contig est égal au nombre de fragments situé à l'extrémité droite d'un contig, montrez que le nombre moyen de contig est N exp(-A)
Les graphes donnant la proportion du génome séquencé et le nombre moyen de contigs sont donné ci-dessous. Vous pouvez modifier les paramètres concernant la longueur G du génome et la longueur L des fragments.
Quelles sont les limites quand A croit de chacun de ces graphes?
Pour le deuxième graphe cette limite n'est pas la limite attendue de 1 seul contig recouvrant tout le génome. Ceci résulte de l'approximation que nous avons faite en négligeant les extrémités.