| BCB 2004 Second Bertinoro Computational Biology Meeting 12-19 June 2004 University of Bologna Residential Center Bertinoro (Forlì), Italy |
 |
|---|
| BCB 2004 Second Bertinoro Computational Biology Meeting 12-19 June 2004 University of Bologna Residential Center Bertinoro (Forlì), Italy |
|
|---|
This year's theme will be Biological Networks: Reconstruction, Analysis, Evolution. In the tradition of the First Bertinoro Computational Biology Meeting, invited speakers will present new results in an environment that promotes informal, interdisciplinary discussion. We expect a broad mix of participants from the biological, computational and mathematical sciences.
Besides the speakers, a small number of PhD students are expected to participate. An expression of interest can be sent to Alessandro Panconesi.
Fellowships: A certain number of BICI-Unesco(ROSTE) grants are available.
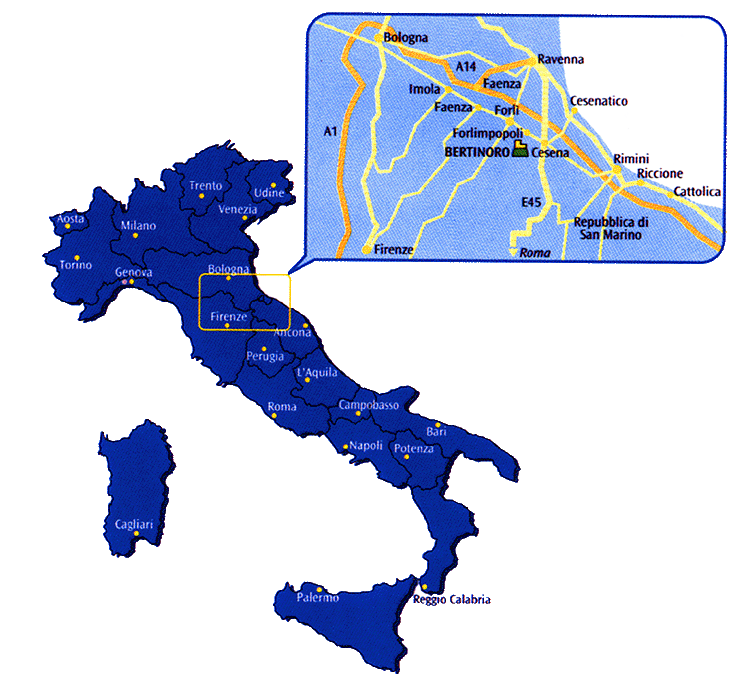
The meeting will be held in the small medieval hilltop town of Bertinoro. This town is in Emilia Romagna about 50km east of Bologna at an elevation of about 230m. Here is a map putting it in context. It is easily reached by train and taxi from Bologna and is close to many splendid Italian locations such as Ravenna and Urbino, treasure troves of byzantine art and history, and the Republic of San Marino (all within 35km) as well as some less well-known locations like the thermal springs of Fratta Terme, the Pieve di San Donato in Polenta and the castle and monastic gardens of Monte Maggio. Bertinoro can also be a base for visiting some of the better-known Italian locations such as Padua, Ferrara, Vicenza, Venice, Florence and Siena.
Bertinoro itself is picturesque, with many narrow streets and walkways winding around the central peak. The meeting will be held in a redoubtable ex-Episcopal fortress that has been converted by the University of Bologna into a modern conference center with computing facilities and Internet access.
From the fortress you can enjoy a beautiful the vista that stretches from the Tuscan Apennines to the Adriatic coast
| 08.00-09.00 | arrivals | breakfast | |||||
|---|---|---|---|---|---|---|---|
| 10.00-10.40 | A. Tramontano | A. Arkin | D. Durand | H. de Jong | |||
| 10.40-11.10 | coffee! | coffee! | coffee! | coffee! | coffee! | coffee! | |
| 11.10-11.50 | J. Stelling | S. Schuster | E. Bornberg-Bauer | C. Ouzounis | L. Farina | D. Thieffry | |
| 12.00-13.30 | lunch! | lunch! | lunch! | lunch! | lunch! | lunch! | |
| 14.00-14.40 | M. Lässig | D. Fell | G. Myers | Sightseeing | T. Przytycka | ||
| 14.40-15.10 | coffee! | coffee! | coffee! | coffee! | |||
| 15.10-15.50 | R. Pinter | I. Koch | D. Kell | D. Pe'er | |||
| 15.50-15.40 | coffee! | coffee! | coffee! | coffee! | departures | ||
| 15.40-16.20 | B. Schwikowski | E. Almaas | A. Valencia | S. Tavazoie | |||
| Arrival: | Saturday 12 June 2004 |
|---|---|
| Departure: | Friday 18 - Saturday 19 June, 2004 |
Cellular metabolism, the integrated interconversion of thousands of metabolic substrates through enzyme-catalyzed biochemical reactions, is the most investigated complex intercellular web of molecular interactions. While the topological organization of individual reactions into metabolic networks is increasingly well understood, the principles governing their global functional utilization under different growth conditions pose many open questions. We implement a flux balance analysis, finding that the network utilization is highly uneven: while most metabolic reactions have small fluxes, the metabolism's activity is dominated by several reactions with very high fluxes. E. coli responds to changes in growth conditions by reorganizing the rates of selected reactions predominantly within this high flux backbone. The identified behavior likely represents a universal feature of metabolic activity in all cells.
Adam ArkinBacterial and animal cells are dynamic machines whose internal chemical networks perform hundreds of complex control and signal processing tasks to govern cellular development over time and in response to deterministic and stochastic signals from the environment. A central challenge in post- genomic biology is to discover the complete physical nature of these networks and to determine if there are principles of control and signal processing by which these cell operate and evolve. If such principles exist then they are handles by which cellular engineers can determine the best placement of external signals (such as pharmaceuticals) to cause a cell to move from an undesired state to a desired state. Here, initial attempts at determining the principles of control, the possible modular structure and the nature of signal flow in cellular networks are briefly introduced. We use examples from bacterial stress response pathways and yeast deletion viability studies to illustrate the principles and approaches.
Erich Bornberg-BauerExplaining the evolution of complexity has been a challenge to Darwinian theory since its conception. At the molecular level, biological complexity involves networks of ligand-protein, protein-protein and protein-nucleic acid interactions in metabolism, signal transduction, gene regulation, protein synthesis and so on. The duplication of genes is the predominant mechanism for the generation of new members of a protein family and so is central to the evolution of complexity. The duplication that increases the size of a network may occur either via single-gene duplication events or by duplication of genes on a large-scale, including the entire genome. The need for networks to remain stable and functional in the cellular environment after the duplication event(s) is thought to favor whole-genome duplication By combining phylogenetic, proteomic and structural information, we have elucidated the evolutionary driving forces for the gene-regulatory interaction networks of bHLH transcription factors. We infer that recurrent events of single-gene duplication and domain rearrangement repeatedly gave rise to distinct networks with almost identical hub-based topologies, and multiple activators and repressors. We thus provide the first empirical evidence for: scale-free protein networks emerging through single-gene duplications, the dominant importance of molecular modularity in the bottom-up construction of complex biological entities, and the convergent evolution of networks.
[1] G. Amoutzias, D.L. Robertson, S.G. Oliver and E. Bornberg-Bauer (2004) Convergent Evolution of Gene Networks by single-gene duplication in higher eukaryotes. EMBO Reports. 5:274-279; (2004).
[2] G. Amoutzias, D.L. Robertson and E. Bornberg-Bauer (2004) Evolution of protein-protein interaction networks in homo- and heterodimerising eukaryotic transcription factors. Comparative and Functional Genomics. 5:79-84.
Dannie DurandTo come soon.
Lorenzo FarinaThe standard paradigm for the analysis and design of engineered control (regulation) systems is the presence of a physical unit called controller able to decide the best action to be taken on the basis of information on the environment provided by sensors. In this perspective, the control unit has a cause/effect structure: the cause being the state of the system as measured by sensors and the effect being the corresponding control action. As noted by Jan Willems [1], in many situations there is no evidence of the existence of a separate control unit, and a goal-oriented behavior simply emerges as the results of specific interconnections. This situation is very common in biochemical networks regulation, where a possible cause/effect structure has to inferred from experimental data and not assumed a priori. In this perspective, one doesn't have to look for sensors and control units, but for behaviors and interconnections [2].
An extraordinary opportunity to uncover behaviors and interconnections in living organisms is offered by the post genomics era [3]. Dissection of transcriptional control can elucidate how metabolism, morphogenesis, differentiation and responses to environment are established, which genes are involved, and, through comparative genomics, how these genes and their networks evolve. In addition, knowledge on transcriptional control of a given process opens the way to develop markers in vivo for remote sensing. A Plant Network Genomics Team has been established aiming to take advantage of new technologies for gene expression analysis in the multicellular model system Arabidopsis thaliana.
[1] J. Willems (1997). On interconnections, control and feedback. IEEE Transactions on Automatic Control 42, 326-339.
[2] J.W. Polderman and J.C. Willems (1998). Introduction to Mathematical Systems Theory: A Behavioral Approach, Springer Verlag, New York, 1998.
[3] C.V. Forst (2002) Network genomics - a novel approach for the analysis of biological systems in the post-genomic era. Mol Biol Rep. 29(3):265-280.
David FellIt would be desirable to be able to build detailed kinetic models of metabolism in order to develop strategies for its modification, for example in drug-based therapies or metabolic engineering. However, the information requirements for this are large, even when an organism has already been subject to extensive biochemical study, and many organisms are now better known from their annotated genomes than from traditional biochemistry and physiology. It is therefore of interest to consider what can be learnt by structural analysis of metabolic networks, since this requires only lists of metabolic and transport reactions occurring in a cell, and these can be obtained from genome annotations. Elementary modes analysis is one approach to the structural study of metabolism that generates all feasible modes of functioning of a network. I will discuss some applications of elementary modes analysis, such as its use for designing modifications to a metabolic network that result in higher yields of a product. Another potential application is as a complement to metabolic flux analysis, where the changing metabolism during a microbial fermentation is interpreted in terms of a changing balance of the modes being utilized by the organism. Other identifiable structural features of metabolic networks are enzyme subsets - groups of enzymes that must always operate together in fixed ratios if they operate at all. It can be hypothesized that these subsets should be apparent in other measures of association, such as occurrence in operons, co-inheritance within phylogenies and co-expression of mRNA and protein. The use of enzyme subsets and related measures as an aid to metabolic interpretation and genome annotation will be illustrated.
Douglas KellNF-kappa-B is a nuclear transcription factor that has been implicated in many cellular processes and disease states (including, apoptosis, arthritis and cancer). We have analysed and extended using Gepasi [1] a model of the NF-kappa-B signalling network [2] controlling the reversible nuclear localisation of the NF-kappa-B molecule. This contains 64 'primitive' reactions and 26 species. Only a few of the reaction parameters exert significant control on the system, but small changes in these parameters can affect the nonlinear dynamics of the system dramatically. This has significant implications both in drug target discovery and for the model building process itself.
[1] Mendes, P. & Kell, D. B. (1998). Non-linear optimization of biochemical pathways: applications to metabolic engineering and parameter estimation. Bioinformatics 14, 869-883.
[2] Hoffmann, A., Levchenko, A., Scott, M. L. & Baltimore, D. (2002). The IkB-NF-kB signaling module: temporal control and selective gene activation. Science 298, 1241-5.
Ina KochBecause biochemical networks tend to be very large and dense, a crucial point is their concise and unambiguous representation and the development of computational methods to model and analyse them in an efficient manner. Before starting a quantitative analysis, a qualitative analysis can check the model for consistency and and correctness of its biological interpretation. For this step we propose methods which are based on Petri nets.
Petri net theory was developed for modelling, analysing, animating, and simulating systems with causal concurrent processes. The theory provides a unique description of complex networks and methods for analysing and simulating them. We have applied these methods to validate metabolic networks [1] as well as signal transduction pathways [2],[3]. The talk will give a short introduction into Petri net basics. Then, model validation of the sucrose breakdown in potato tuber will be explained and discussed in detail. This includes the hierarchical modelling, the calculation of structure properties and analysis of the dynamic behaviour. A special focus will be the determination and discussion of system invariants.
[1] Klaus Voss, Monika Heiner, Ina Koch. Steady State Analysis of Metabolic Pathways using Petri Nets. In Silico Biology 3(3):367-387 (2003).
[2] Monika Heiner, Ina Koch, Jürgen Will. Model Validation of Biological Pathways Using Petri Nets - Demonstrated for Apoptosis. BioSystems, Special issue, in press (2004).
[3] Monika Heiner, Ina Koch. Petri Net Based Model Validation in Systems Biology. LCNS, Proc. of 25th International Conference on Application and Theory of Petri Nets July, 21-25, Bologna, Italy, in press (2004).
Hidde de JongIn order to cope with the large amounts of data that have become available in genomics, mathematical tools for the analysis of networks of interactions between genes, proteins, and other molecules are indispensable [1]. I will present a method for the qualitative simulation of genetic regulatory networks, based on a class of piecewise-linear (PL) differential equations that has been well-studied in mathematical biology [2]. The simulation method is well-adapted to state-of-the-art measurement techniques in genomics, which often provide qualitative and coarse-grained descriptions of genetic regulatory networks. Given a qualitative model of a genetic regulatory network, consisting of a system of PL differential equations and inequality constraints on the parameter values, the method produces a graph of qualitative states and transitions between qualitative states, summarizing the qualitative dynamics of the system. The qualitative simulation method has been implemented in Java in the computer tool GNA (Genetic Network Analyzer, available at http://www-helix.inrialpes.fr/gna) [3]. I will discuss the application of the computer tool to the modeling and simulation of several bacterial regulatory systems, in particular the networks controlling the initiation of sporulation in Bacillus subtilis and the nutritional stress response in Escherichia coli [4].
[1] H. de Jong (2002), Modeling and simulation of genetic regulatory systems: A literature review, Journal of Computational Biology 9(1):69-105.
[2] H. de Jong, J.-L. Gouzé, C. Hernandez, M. Page, T. Sari, J. Geiselmann (2004), Qualitative simulation of genetic regulatory networks using piecewise-linear models, Bulletin of Mathematical Biology, 66(2):301-340.
[3] H. de Jong, J. Geiselmann, C. Hernandez, M. Page (2003), Genetic Network Analyzer: Qualitative simulation of genetic regulatory networks, Bioinformatics, 19(3):336-344.
[4] H. de Jong, J. Geiselmann, G. Batt, C. Hernandez, M. Page (2004), Qualitative simulation of the initiation of sporulation in Bacillus subtilis, Bulletin of Mathematical Biology, 66(2):261-300.
Michael LässigThe genomes of higher organisms are highly collective systems with multiple interactions between genes. These genetic networks are linked to other levels of molecular information processing such as protein interaction maps or metabolic networks. Their combinatorial complexity is an essential characteristic of higher organisms, allowing a large number of functional tasks to be performed by a limited number of genes. This talk describes our current understanding of the structure and evolution of molecular networks. Particular emphasis is paid to the links between the statistics of regulatory sequences and functional patterns in gene networks.
Gene MyersWe discuss the possibility of a program of high-throughput in-situ image analysis in D. melanogaster and C. elegans embryos. We describe what information we might collect and what we might be able to infer form it. It is our contention that this may be the best way to understand development from a systems perspective.
Christos OuzounisTo come soon.
Dana Pe'erModularity is emerging as an organizing principle in biological systems, including transcriptional networks, signal transduction systems and metabolic pathways. The detailed identification and characterization of individual modules traditionally depends on intensive experimental studies. In our work, we develop computational learning methods that use high throughput genomics data to automatically and simultaneously identify the components, control and behavior of biological modules. We take a model based approach, and build an abstraction of the biological system that can generate predictions on the systems behavior under different condition. We define a class of possible models and use computational learning procedures to select the model that best explains the measured observations. In particular, we focus on inferring the regulation of gene expression. We develop and learn a series of models of increasing complexity and accuracy of structure and regulation program. We show, with extensive statistical, bioinformatics, and biological validations, that complex combinatorial regulation can be accurately inferred solely and directly from gene expression measurements. As more diverse genome wide datasets become available in various organisms, it is our belief that our model based approach may result in important new insights in the ongoing endeavor to understand the complex web of biological regulation.
Ron PinterSeveral genome-scale efforts are underway to reconstruct metabolic networks for a variety of organisms. As the resulting data accumulates, the need for analysis tools increases. A notable requirement is a pathway alignment finder that enables both the detection of conserved metabolic pathways among different species as well as divergent metabolic pathways within a species. When comparing two pathways, the tool should be powerful enough to take into account both the pathway topology as well as the nodes? labels (e.g. the enzymes they denote), and allow flexibility by matching similar - rather than identical - pathways.
MetaPathwayHunter is a pathway alignment tool that, given a query pathway and a collection of pathways, finds and reports all approximate occurrences of the query in the collection, ranked by similarity and statistical significance. It is based on novel, efficient graph matching algorithms that extend the functionality of known techniques. Our program also supports a visualization interface with which the alignment of two homologous pathways can be graphically displayed.
We employed this tool to study the similarities and differences in the metabolic networks of several organisms (as represented in highly curated databases that are available on the World Wide Web). We reaffirmed that most known metabolic pathways common to bacteria and yeast are conserved; furthermore, we present a few cases in which the comparison of metabolic pathways between species exemplifies divergent and putative convergent evolution, and within a species - exemplifies divergent evolution. We conclude with a description of several biologically meaningful meta-queries, demonstrating the power and flexibility of our new tool in the analysis of metabolic pathways.
Teresa PrzytyckaRecent studies of properties of various biological networks revealed that many of them display scale free characteristics. Since the theory of scale free networks is applicable to evolving networks, one can hope that it provides not only a model of a biological network in its current state but also sheds some insight into the evolution of the network. But how well are the characteristics of scale free theory reflected in biological networks? To answer this question, we investigate the probability distributions and scaling properties underlying some models for biological networks and protein domain evolution. In particular, we have examined the recently proposed "big-bang" (BB) protein domain evolution model and a hierarchical preferential attachment (HPA) model that we designed here for the illustrative purpose. Most importantly, the BB model includes evolutionary drift while HPA doesn't. We analyzed the computer-generated data from the BB model and the HPA model. Although at short time scale both the BB model and the HPA model seem to follow the pattern expected from the scale free theory, they diverge as the length of the simulation increases. Basically, with the long time scale results included, the HPA model follows scale free distribution more closely while the BB model is much better fitted by a Yule distribution. We examine the reasons and possible consequences of this finding.
Stefan SchusterThe study of competitive and cooperative strategies in the interaction between different microbial species is of potential relevance for the optimization of biotechnological setups. Two species (or strains) of micro-organisms that use the same nutrient, but may choose between two different pathways of ATP production, are studied from a game-theoretical point of view. The pathways are considered as distinct strategies to which payoffs can be assigned. The payoffs are assumed to be proportional to the steady-state number of individuals sustainable on the basis of these strategies. For each species (or strain), this numbers does not only depend on the strategy chosen by that species but also on the strategy of the other species. In a certain parameter range, the payoffs fulfil the conditions for the prisoner's dilemma. Therefore, cooperative behaviour is unlikely to occur, unless additional factors interfere. In fact, the yeast Saccharomyces cerevisiae uses a competitive strategy by fermenting sugars even under aerobic conditions, thus wasting its own resource. The simple quantifiable structure of the model should enable one to experimentally determine a payoff matrix. Several ideas generalizing the above results are discussed, in particular, with respect to possible scenarios of transition to cooperative behaviour.
Benno SchwikowskiMany protein-protein interactions are mediated by peptide recognition modules (PRMs), compact domains that bind to short peptide motifs, and play a critical role in many biological processes. It is known that subtle variations in each motif instance confer a high degree of selectivity to the induced interactions. Existing approaches to derive the motifs computationally fail, because modeling each individual motif instance provides too little data to the learning process, and simple global models are unable to account for the observed specificity.
On the basis of an all vs. all dataset recently published for the SH3
PRM (Tong et al., 2002, Science
Jörg Stelling
Understanding biological networks: Which knowledge and data are needed?
The new `omics technologies have fueled the interest in understanding complex biological networks through a combination of comprehensive measurements and (quantitative) mathematical modeling. At present, however, it is largely unclear, which knowledge and data will be required for establishing realistic mathematical models, and, perhaps equally important, to what extent the already available data allow for meaningful model development. The talk will address these questions by relying on two examples: structural analysis of metabolic networks and dynamic analysis of a complex network in yeast cell cycle regulation. Both cases demonstrate an unexpectedly high degree of information that one can extract from the available data, and suggest strategies for efficiently linking experimental and theoretical approaches to cellular networks.
Saeed TavazoieI will discuss the challenge of learning regulatory networks from whole-genome observations of gene expression and DNA sequences. I will show that a sufficiently general probabilistic framework, constrained by mechanistic insights, can capture a large fraction of regulatory information in DNA. This approach generates complex combinatorial rules, which allow successful prediction of gene expression patterns from sequence alone.
Denis ThieffryA proper understanding of the mechanisms controlling gene expression requires the integration of molecular and genetic data into full fledge mathematical models. An overview of the main dynamical modelling approach will be provided, before focusing on a multi-level, logical approach, which enables a flexible qualitative modelling of complex regulatory networks. This approach encompasses the development of a dedicated software suite (GIN-sim), and will be illustrated by applications to cell cycle and cell differentiation in model animals.
Anna TramontanoTo come soon.
Alfonso ValenciaCurrent High Throughput experimental techniques are providing increasingly complex data in areas such as: genome sequences, expression arrays, yeast two-hybrid (y2h), TAPs/MS and peptide libraries. Bioinformatics and Computational Biology play a crucial role in the organization and analysis of this information. During this presentation I'll describe the current computational approaches for the prediction of interaction partners based on sequence information. I will particularly focus on the methods that my group has developed, and the comparison with other computational and experimental approaches (see Pazos & Valencia, Curr. Opinions in Struc. Biol., 2002, Hoffmann Valencia TIGs 2004).
The natural consequence of these applications in Biology is the extension of the predictions to the molecular level, helping in the understanding of the molecular basis of the corresponding protein interactions. I'll present our view on how the methods developed for the description of protein interaction networks can be tailored for the prediction of the molecular features of individual interactions (protein docking), and protein functional sites (del Sol et al., J.Mol.Biol 2003).
Finally, interactions can be also obtained from the many detailed experiments carried out by individual laboratories and published in the scientific literature. The development of Information Extraction technology has become one of the more exciting new fields of Bioinformatics. The main challenges include: i) access and organization of the textual information (document retrieval); ii) development of comprehensive repositories of annotated text in the various knowledge domains; iii) identification of entities in text, particularly protein and gene names, but also diseases, drugs, species, tissues and others; iv) accurate description of the relations between entities at the level of pair wise relations (e.g. protein interactions and gene control relations), relations between entities (e.g. genes associated to a given disease), and at the level of global relations (e.g. functions common to a set of genes), and v) representation of the extracted knowledge, including technical issues (e.g. graphical formats, database querying capabilities), scoring and summarizing the information extracted from text. For a review see, Blaschke et al., Brief. Bioinfor. 2002 and Blaschke, Valencia, EEEI, 2002.
In response to these challenges it is very important to compare and assess competing approaches with common standards and evaluation criteria (see the description of the BioCreative competition at http://www.pdg.cnb.uam.es/BioLINK/BioCreative.eval.htm). I'll review the situation of the field at the light of the results of the BioCreative assessment.
Finally, integrating all these information: experimental and predicted interaction maps, detailed predictions of binding surfaces with the information collected from databases and scientific literature, represents a formidable task of Integration of heterogeneous data, that requires a serious assessment of the available software engineering, data management and web technologies.
| Scientific Organizing Committee | Dannie Durand, Carnegie Mellon University, Pittsburgh, USA |
|---|---|
| Anna Tramontano, University of Rome "La Sapienza", Rome, Italy | |
| Marie-France Sagot, Inria Rhône-Alpes and University Claude Bernard, Lyon, France | |
| Local Organization | |
| Andrea Bandini, Elena Della Godenza, Centro Congressi di Bertinoro | |
| Sponsored by |
|
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}