|
Biological Networks: Interaction with Genome and
Developmental Evolution 21-28 May 2005 University of Bologna Residential Center Bertinoro (Forlì), Italy |
 |
|---|
|
Biological Networks: Interaction with Genome and
Developmental Evolution 21-28 May 2005 University of Bologna Residential Center Bertinoro (Forlì), Italy |
|
|---|
In 2004, the meeting on Biological Networks: Reconstruction, Analysis, Evolution brought together scientists from diverse backgrounds and perspectives for a free and exciting exchange of ideas. It was a great success: excellent talks, good food and a relaxed atmosphere, all in a beautiful location. We have every expectation that the 2005 meeting on Biological Networks: Interaction with Genome Evolution will be just as exciting. Following the tradition established in the first meeting, invited speakers will present new results in an environment that will promote lively, synergistic discussions of novel ideas on the computational analysis of biological networks, genome structure and dynamics.
Besides the speakers, a number of PhD students are expected to participate and present their work to a small and friendly public. A call for posters will consequently be made later.
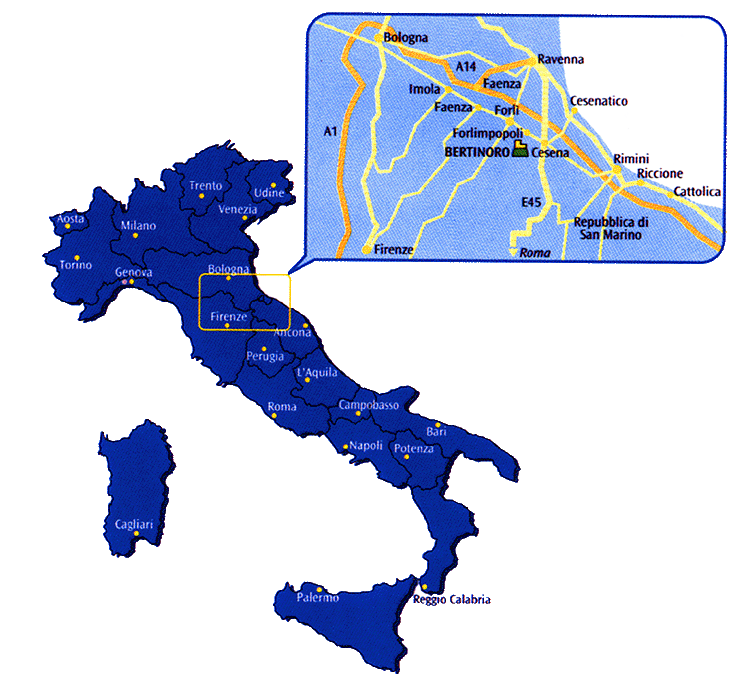
The meeting will be held in the small medieval hilltop town of Bertinoro. This town is in Emilia Romagna about 50km east of Bologna at an elevation of about 230m. Here is a map putting it in context. It is easily reached by train and taxi from Bologna and is close to many splendid Italian locations such as Ravenna and Urbino, treasure troves of byzantine art and history, and the Republic of San Marino (all within 35km) as well as some less well-known locations like the thermal springs of Fratta Terme, the Pieve di San Donato in Polenta and the castle and monastic gardens of Monte Maggio. Bertinoro can also be a base for visiting some of the better-known Italian locations such as Padua, Ferrara, Vicenza, Venice, Florence and Siena.
Bertinoro itself is picturesque, with many narrow streets and walkways winding around the central peak. The meeting will be held in a redoubtable ex-Episcopal fortress that has been converted by the University of Bologna into a modern conference center with computing facilities and Internet access.
From the fortress you can enjoy a beautiful the vista that stretches from the Tuscan Apennines to the Adriatic coast
Attention: This is still subject to some small changes.
| 08.00-09.30 | arrivals | breakfast | |||||
|---|---|---|---|---|---|---|---|
| 10.00-10.40 | L. Duret | I. Wapinski | D. Wolf | S. Miyano | J. Papin | Open problem 3 | |
| 10.40-11.10 | coffee! | coffee! | coffee! | coffee! | coffee! | coffee! | |
| 11.10-11.50 | A. Ruiz | D. Segrè | H. Meinhardt | I. Koch | J. Pereira-Leal | Open problem 3 | |
| 12.00-13.30 | lunch! | lunch! | lunch! | lunch! | lunch! | lunch! | |
| 14.00-14.40 | E. Alm | M. Singh | R. Sommer | sightseeing | M. Samoilov | departures | |
| 14.40-15.10 | coffee! | coffee! | coffee! | coffee! | |||
| 15.10-15.50 | M. Chandler | D. Durand | E. Siggia | K. Kohn | |||
| 15.50-16.20 | coffee! | coffee! | coffee! | coffee! | |||
| 16.20-17.00 | M. Dunham | T. Przytycka | F. Naef | J. F. Poyatos | |||
| 17.00-18.00 | Open problem 1 | Open problem 2 | |||||
Open problem sessions are meant for free discussions among the participants on a number of topics. This year, since the topics covered by the meeting are broader than last year, we wish to have more formally organized sessions. In particular:
Open problem session 3 is a more general affair and will have a freer format. It has for main objective enabling participants to discuss in more detail of possible formal or informal future collaborations. Discussion on potential collaborations will of course also be possible at the other open sessions.
There will be one person chairing each Open problem session but all participants are strongly encouraged to prepare topics to be discussed in agreement with the spirit of each session.
| Arrival: | Saturday 21 May, 2005 |
|---|---|
| Departure: | Friday 27 - Saturday 28 May, 2005 |
Registration is now open here.
Eric Alm
Operon Formation is Driven by Co-regulation and Not by Horizontal Gene
Transfer
Background:
The organization of bacterial genes into operons was
originally ascribed to the benefits of co-regulation [1]. More
recently, the "selfish operon" model was proposed, in which new
operons are formed by repeated gain and loss of genes
[2]. Empirically, operons often appear to be subject to horizontal
gene transfer (HGT), yet non-HGT genes are particularly likely to be
in operons [3]. To clarify whether HGT is involved in the formation of
new operons or simply the propagation of pre-existing operons, we
identified and studied a comprehensive set of recently formed operons
in Escherichia coli K12.
Results:
Genes that have homologs in distantly related bacteria
but not in close relatives of E. coli (indicating likely HGT)
form new operons at about the same rates as native genes. Furthermore,
genes in new operons are no more likely than other genes to have
phylogenetic trees that are inconsistent with the species tree. In
contrast, essential genes and ubiquitous genes without paralogs
(believed to undergo HGT rarely) often form new operons. These results
argue that HGT is not a cause of operon formation, but instead helps
to propagate pre-existing operons to new genomes.
To identify other factors that may play a role in operon formation, we hypothesized that in cases where gene regulation is complex, forming operons by gene rearrangement might be easier than evolving complicated promoter sequences multiple times independently. By contrast, if little regulatory information is required to specify optimal expression patterns, then independent promoters might be favored. To test this hypothesis, we used phylogenetic footprints reported in previous studies of bacterial transcriptional regulation as an estimate of regulatory complexity [4, 5]. Consistent with our hypothesis, genes whose upstream regions had a greater number of base pairs of DNA falling into phylogenetically conserved "footprint" regions were more likely to occur in operons.
Conclusions:
Operons are ubiquitous and are often the unit of
horizontal gene transfer. While HGT plays a key role in the
propagation of operons (nearly 50% of HGT genes in E. coli were
found to be part of horizontally transferred operons), it does not
seem to be crucial to the formation of new operons. Finally, we find
that in cases where gene regulation is complex, forming operons
provides an easier solution to the problem of co-regulation than
evolving independent promoters.
References:
1. F.a.M. J. Jacob, Cold Spring Harbor Symp. Quant. Biol. 26:
193-211, 1961.
2. J.G. Lawrence, and J.R. Roth, Selfish operons: horizontal transfer
may drive the evolution of gene clusters. Genetics, 143(4):
1843-1860, 1996.
3. C. Pal and L.D. Hurst, Evidence against the selfish operon theory.
Trends Genet, 20(6): 232-4, 2004.
4. L. McCue et al., Phylogenetic footprinting of transcription factor
binding sites in proteobacterial genomes. Nucleic Acids Res,
29(3): p. 774-82, 2001.
5. G. Terai, T. Takagi, and K. Nakai, Prediction of co-regulated genes
in Bacillus subtilis on the basis of upstream elements conserved across
three closely related species. Genome Biol, 2(11): 1-12, 2001.
Maitreya Dunham
Genome Evolution in Saccharomyces Yeasts
The Saccharomyces clade of yeasts provides a wonderful system for studying genome evolution at many scales. My lab works on the shorter timeframes of experimental evolution, using DNA microarrays to interrogate the genome of "evolved" yeast. Over just a few hundred generations of nutrient-limited growth in chemostats, yeast cultures accumulate changes in copy number ranging from amplification of a single transporter gene to aneuploidy for entire chromosomes. We are attempting to characterize and understand the consequences of these changes in the genome. The breakpoints of these rearrangements are almost always associated with repetitive genome elements, including transposons, transposon fragments, duplicated genes, and regions of microhomology. A series of studies from other groups, plus further work in my own lab, has emphasized the generality of our results. Modern Saccharomyces yeasts' genomes are the result of a whole genome duplication followed by massive gene loss, making gene duplication a driving factor in yeast evolution. Comparison of the different sensu stricto species reveals a number of genome rearrangements with repeated elements as their breakpoints. In S. cerevisiae, other screens for chromosome rearrangements have found identical breakpoints to those in our studies, including particularly fragile sites. Wild yeasts also commonly harbor rearrangements. Experimental evolution of wild strains recapitulated at least one rearrangement commonly found in natural isolates. Together these results demonstrate some of the fundamental aspects of yeast evolution.
Dannie Durand
Deciphering multi-domain evolution: Exploiting structure
in the protein domain network
Joint work with Teresa Przytycka at NIH.
Protein evolution through duplication and rearrangement of functional subunits called domains is Nature's equivalent of rapid prototyping. Genomic evidence suggests that the functional possibilities offered by the combinatorial explosion of domain arrangements represented a special evolutionary opportunity. Multi-domain proteins played a key role in the evolution of multicellularity and are prevalent in the cell-cell interactions, tissue repair, cell death and the immune system. In this talk, I will present a computational approach to investigating the forces drive the formation of these modular sequences. We model the sequence universe as a graph and show how structural properties of this graph can be used to investigate the evolutionary processes that engendered them.
Laurent Duret
Relationships between genome organization and gene expression in mammals
Joint work with Marie Sémon.
Gene order in mammalian genomes is not random: co-expressed genes, and notably housekeeping genes, are significantly more clustered than expected by chance. This clustering either might reflect a selective pressure to group co-expressed genes into transcription-competent chromatin domains, or might be a neutral consequence of interferences between the transcriptional activity of neighbor genes. To test both models, we analyzed the frequency of chromosomal breakages (inferred by comparison of human and chicken genomes) within or outside clusters of co-expressed genes in human, and the evolution of the expression patterns of linked genes in human and mouse. (i) Analysis of chromosomal breakages shows that linkage is retained more frequently for genes that belong to the same co-expression cluster in human: some clusters of co-expressed genes are maintained by natural selection. This effect is however relatively weak since these clusters include at most 5% of the mammalian genes. (ii) Analysis of expression evolution between human and mouse shows that the expression change of a gene is linked to the evolution of its neighbors. We observed these «interferences» only between genes closely located on the genome. We built a simple model to estimate the probability that the expression change of a gene propagates to its neighbors. We found a weak but significant probability (2.5%). Using simulations, we show that this interference effect is strong enough to generate many of the observed clusters in the human genome. We propose that both selective and neutral evolutionary processes contribute to the non-randomness of gene order with respect to their expression pattern in human.
Ina Koch
Qualitative analysis of signal transduction pathways - considering as
example the pheromone pathway in S. cerevisiae Yeasts
Joint work with Andrea Sackmann and Monika Heiner.
To answer many evolutionary questions we have to get knowledge of gene expression control and gene interactions. Signal transduction - also called information metabolism - represents a mechanism of the cell to control gene regulation.
Petri net modelling tools and analysis techniques provide sound methods to investigate biochemical models qualitatively as well as quantitatively. We suggest before starting the quantitative modelling to develop a basic qualitative model which is validated by qualitative analysis techniques based on Petri net theory. We have applied this approach successfully to metabolic networks. Modelling signal transduction pathways certain properties have to be considered. The pheromone pathway in S. cerevisiae is well investigated and serves as a model system for eukaryotes. We have modelled this pathway qualitatively as a P/T Petri net and analysed the static and dynamic properties of the net. The result is an comprehensive model of the pheromone pathway in yeast.
In the talk we give first a short introduction to Petri nets for an intuitive understanding. We explain the model of the pheromone pathway in yeast in detail focusing on special properties concerning signal transduction pathways.
Hans Meinhardt
Models for pattern formation and primary axes formation in higher organisms
A crucial step in early the development of higher organisms is the establishment of the primary body axes, anteroposterior (head-to tail) and dorsoventral (back-to belly). Simpler radial-symmetric animals such as the freshwater polyp hydra have only a single axis that is assumed to be ancestral. Models will be discussed that account for this ancestral pattern formation and for the transition to bilateral symmetry. In present-day higher organisms the ancestral patterning system accomplishes the basic subdivision in the developing brain, suggesting that during evolution the ancestral body pattern evolved into the brain of higher organisms. The trunk, the largest part of higher organisms, is an evolutionary later addition. The patterning of the trunk follows different rules and is frequently connected with the formation of segments. Models for the formation and evolution of segments will be discussed. Bilaterality is proposed to have its origin in the mutual reorientation of two pattern- forming system that existed already in the radial-symmetric ancestors. Patterning along the dorsoventral axis of an animal with a long-extended head-to-tail axis requires the formation of stripe-like organizing regions that extend along the whole body axis. It will be shown that the formation such a midline organizer is an intricate patterning problem that requires the coupling of several pattern-forming reactions. In vertebrates and insects different mechanisms are realized for the establishment of the midline organizer. These models provide a framework for the evolution of the different modes of body axes formation.
Satoru Miyano
Gene Networks for Systems Biology
Gene networks play a central role in systems biology. This talk presents two computational approaches related to gene networks.
Firstly, computational methods for estimating gene networks from microarray gene expression data are presented. We consider microarray data obtained by various perturbations such as such as gene disruptions, shocks, drug responses, time-course measurements, etc. The idea is to combine the Bayesian network approach with nonparametric regression, where genes are regarded as random variables and the nonparametric regression enables us to capture from linear to nonlinear structures between genes. As a criterion for choosing good networks, we defined a kind of information criteria called the BNRC (Bayesian network and Nonparametric Regression Criterion) score. Naturally, the sole use of microarray data has limitations on gene network estimation. For improving the biological accuracy of estimated gene networks, we have made a general framework by extending this method so that it can employ genome-wide other biological information such as sequence information on promoter regions, protein-protein interactions, protein-DNA interactions, subcelluar localization information, and literature. The problem of finding an optimal Bayesian network is known computationally intractable. We also developed an algorithm for searching and enumerating optimal and suboptimal Bayesian networks in feasible time on supercomputers. Computational experiments with this search algorithm have provided evidences of the biological rationality of our computational strategy.
Secondly, a software tool for modeling and simulating gene networks which is based on the notion of Petri net is presented. Obviously, an important challenge is to create a software platform with which scientists in biology/medicine can comfortably model and simulate dynamic causal interactions and processes in the cell(s) such as gene regulations, metabolic pathways, signal transduction cascades, etc. For this direction, we defined a notion called Hybrid Functional Petri Net with extension (HFPNe). It was implemented in our software Cell Illustrator (http://www.gene-networks.com/). Since Cell Illustrator equips a biology- oriented GUI, modeling of very complex biological processes with HFPNe can be performed in a simply way. Its effectiveness was demonstrated by modeling biological processes such as alternative splicing, frameshifting, Huntington's disease model, p53 modifications.
Felix Naef
Interaction between two clocks in populations of circadian
oscillators
We model cellular networks of circadian oscillators to interpret recordings of a luciferase reporter in a circadian cell culture assay. Correlation with single cell data illustrates the complimentary of both techniques. Our analysis uncovered reciprocal interactions between the circadian and cell cycle oscillators, manifest for example as a gating of mitosis time by the clock.
Jason Papin
From intracellular signaling networks to multicellular function
The reconstruction and mathematical analysis of cellular signaling networks is a pressing challenge. Large stoichiometric reconstructions of signaling networks account for the interconnectivity and functional relationships among numerous cellular components. With these detailed reconstructions, mathematical analysis techniques can describe network properties and their relationships to disease states. I will present methods for reconstructing and analyzing large cellular signaling networks as well as results from a study of the JAK-STAT signaling network in the human B-cell. With the JAK- STAT signaling network as an example, I will describe mathematically-based network properties including input-output relationships, correlated reaction sets (or unbiased network modules), and network crosstalk. Current efforts to integrate multiple scales of biological networks (from intracellular events to multicellular function) will also be discussed.
José B. Pereira-Leal
The origins of modularity in cellular networks
Modularity is an attribute of a system that can be decomposed into a set of cohesive and loosely coupled modules. The notion of modularity is also familiar in biology. For example proteins are composed of structural domains, which are evolutionary modules and the building blocks of proteins. These are reused and combined in different ways, thus facilitating the generation of complexity as well as structural and functional diversity. At higher levels of biological organization modularity is also prevalent, in particular in cellular networks. The evolutionary mechanisms driving the evolution of modularity in networks are poorly understood. Although we know that gene duplication is the major force driving genome evolution, it is unclear to what extent it contributes to the emergence and maintenance of modularity. In this talk I will discuss recent results on the evolution of protein interaction networks and protein complexes, which are functional modules in these networks. I will argue that modularity is a simple consequence of gene duplication with conservation of interactions. While duplication mostly results in network growth, one particular class of interactions underlies the formation of functional modules in these networks.
Juan F. Poyatos
Genome architecture and the organization of cellular networks: Evidence
of coevolution in yeast
Joint work with Lawrence D. Hurst
Which are the factors contributing to the complex distribution of genes around eukaryotic genomes? Two models have been recently considered. One suggests that genes are clustered to favour their co-regulation. An alternative classic model argues that genes are clustered to reduce their recombination rate. I will discuss a systematic analysis of this problem and a possible solution. This solution, using the yeast Saccharomyces cerevisiae as model organism, shows the relationship between genome architecture and the organization of the protein interaction networks and it is consistent with a model for eternal cycling of gene order evolution.
Teresa Przytycka
Network motifs and evolution of hard to gain and hard to loose character
traits
Recent studies of properties of various biological networks have been focusing on discovering characteristic features of such networks. Following the discovery that degree distribution in biological networks is typically well approximated by scale free distribution, researchers started to look for other network measurements that would be more powerful in discriminating between various types of networks. Such measurements include, for example, clustering coefficient, diameter, and most recently network motifs. Once over-represented and under-represented network motifs are identified, the next challenge is to understand the biophysical and/or evolutionary roots of a given network topology.
In this talk we study the networks associated with hard to gain and hard to loose characters, as for example introns. In this talk we first study characteristic network motif signature of such a network and then we provide a theoretical argument that proves that these motifs are to be expected in these networks and are in fact a consequence of a more global topological property.
Alfredo Ruiz
Conservation of regulatory sequences and gene expression patterns in
the disintegrating Drosophila Hox gene complex
Joint work with Bárbara Negre, Sònia Casillas, Magali Suzanne, Ernesto Sánchez-Herrero, Michael Akam, Michael Nefedov, Antonio Barbadilla and Pieter de Jong.
Homeotic (Hox) genes are usually clustered and arranged in the same order as they are expressed along the anteroposterior body axis of metazoans. The mechanistic explanation for this colinearity has been elusive and it may well be that a single and universal cause does not exist. The Hox gene complex (HOM-C) has been rearranged differently in several Drosophila species producing a striking diversity of Hox gene organizations. We investigated the genomic and functional consequences of the two HOM-C splits present in Drosophila buzzatii. Firstly, we sequenced two regions of the D. buzzatii genome, one containing the genes labial and abdominal A, and another one including proboscipedia, and compared their organization to that of D. melanogaster and D. pseudoobscura in order to map precisely the two splits. Then a plethora of conserved non-coding sequences, which are putative enhancers, were identified around the three Hox genes closer to the splits. The position and order of these enhancers are conserved, with minor exceptions, between the three Drosophila species. Finally, we analyzed the expression patterns of the same three genes in embryos and imaginal discs of four Drosophila species with different Hox gene organizations. The results show that their expression patterns are conserved despite the HOM-C splits. We conclude that, in Drosophila, Hox gene clustering is not an absolute requirement for proper function. Rather, the organization of Hox genes is modular and their clustering seems the result of phylogenetic inertia more than functional necessity.
Michael Samoilov
Role of Non-Classical Dynamics in the Analysis of Biological Network
Function
The non-deterministic nature of reactions underlying the gene- and proteom- regulatory biological networks has now been broadly accepted, with the chemical master equation (CME) becoming the de facto basis for its accurate representation (1). However, while much fruitful discussion has been dedicated to, among other things, studying stochastic effects during gene expression (2, 3), the questions of general role and relevance of non- classical regimes for in situ biomolecular systems remain largely unresolved. Moreover, it has frequently been perceived that - relative to the classical deterministic chemical kinetics (CCK) - the non-classical mechanisms either contribute only marginally to the overall system dynamics or else require very small numbers of molecules and substantial reaction complexity to manifest themselves. This, however, turns out not to be the case. In fact, biological motifs as basic and ubiquitous as futile cycles have been shown not only to be capable of exhibiting characteristic behaviors qualitatively different from those expected classically (under CCK) at high molecular counts, but possibly of offering new functional modalities to any biological network they are imbedded in (4).
Thus the question naturally arises as to the general conditions required for molecular reaction systems to be able to show substantial behavioral deviations from CCK and whether such conditions are indeed present in natural molecular systems. If these non-classical behavioral modes were veritably available, but not encountered anywhere in a biological network - this would present an evolutionary puzzle, since such functional dynamics offer an organism additional opportunities for diversification without extracting much if anything in terms of energetic or entropic costs. Alternatively, if they are actually present somewhere - the up and downstream propagation of local effects would generally imply the need to include non-classical CME dynamics in the analysis of any underlying biological network function. Based on the complete CME formalism and some auxiliary methods, this talk will consider such issues in the context of basic reaction stoichiometry.
References:
1. Gillespie, D. T., Markov processes. Academic Press,
Boston), 1992.
2. McAdams, H. H. & Arkin, A., Proc Natl Acad Sci U S A,
94:814-819, 2007.
3. Elowitz, M. B., Levine, A. J., Siggia, E. D. & Swain, P. S.,
Science, 297:1183-6, 2002.
4. Samoilov, M., Plyasunov, S. & Arkin, A. P., Proc
Natl Acad Sci U S A, 102:2310-2315, 2005.
Daniel Segrè
Optimality and modularity in metabolic networks
Understanding how cellular metabolism depends on the functions and interactions of single genes and pathways constitutes a major challenge, which requires simplifying assumptions. In flux balance models [1], metabolic networks are treated as steady state systems, whose reaction rates (fluxes) can span a space of solutions constrained by fundamental mass conservation laws. Efficient optimization algorithms can search this space for flux arrangements that optimize a given objective function, such as cellular growth. Using this approach, we performed large-scale computer experiments of single and double gene deletions. Through the comparison of model predictions and experimental data for single gene deletions in E. coli, we could gain new insight about the cellular response to metabolic perturbations. In particular, we found that the perturbed networks can deviate significantly from optimal growth capacity, and are better described by assuming a minimal redistribution of fluxes with respect to the unperturbed cell [2].
The study of double gene deletions allowed us to gain insight about the functional organization and modular structure of biological networks. Double gene perturbations uncover epistatic interactions, manifested in the way mutations affect each other's phenotypes. In a large scale analysis of the yeast S. cerevisiae we identified pairs of genes whose combined deletion affects growth much more (synergistic), or much less (buffering) than what expected based on the single deletions. We found that the network of these epistatic interactions can be organized hierarchically into function-enriched modules that interact "monochromatically," i.e. with purely synergistic or purely buffering links [3]. This property extends epistasis from single genes to functional units, and provides a novel definition of biological modularity, which emphasizes interactions between, rather than within, functional modules.
References:
[1] Covert MW, Schilling CH, Famili I, Edwards JS, Goryanin II, Selkov
E, Palsson BO, Metabolic modeling of microbial strains in silico,
Trends Biochem Sci. 26(3):179-186, 2001.
[2] Segrè D, Vitkup D, Church GM, Analysis of optimality in natural
and perturbed metabolic networks, Proc Natl Acad Sci U S A.
99(23):15112-15117, 2002.
[3] Segrè D, Deluna A, Church GM, Kishony R, Modular epistasis in
yeast metabolism, Nat Genet. 37(1):77-83, 2005.
Eric Siggia
Computation approaches to blastoderm patterning in the fly and its
evolution
Using prior information about the binding preferences of transcription factors that define the anterior-posterior patterning system in flies we are able to locate most of the cis regulatory modules (CRM) implicated in this process and validate our results by experiment. The recently completed D.pseudoobscura genome, provides the means for a comprehensive assessment of stasis and change in regulatory DNA. We have analyzed ~100 known and putative cis regulatory modules that control the key genes involved in AP patterning.
We did a computational screen on the CRM's to isolate a subset for
experimental study that best exemplified the following patterns of
molecular evolution:
(1) large (100's bp) indels with plausible binding sites;
(2) systematic loss of binding sites for specific factors.
Even in the absence of large indels, the computation classifies
homologous modules as:
(3) synergistic (the two species score S2 is the sum of the single
species scores), or
(4) antagonistic (S2 no larger than the maximum single species score).
In the former case two species aids in the identification of CRM,
while in the latter case it does not. The consequences of these
changes will be illustrated by experiments.
When considering the entire regulatory region of a gene, there are
instances of:
(5) functionally duplicate modules, which generate overlapping subsets
of the native pattern and;
(6) pseudo modules; regions with many AP factor binding sites, yet
lacking a component (e.g. activator) required for
functionality. Overall several of the themes known from the
evolution of the proteome are recapitulated by the regulatory
sequence.
Mona Singh
Predicting and analyzing protein interaction networks
Abstract soon available.
Ralf J. Sommer
The evolution of nematode vulva development - what we can learn from a
case study
Cell fate specification and cell-cell signaling have been well studied during Caenorhabditis elegans vulva development and provide a paradigm in evolutionary developmental biology. We have developed the nematode Pristionchus pacificus as a satellite organism with an integrated physical and genetic map that allows detailed comparisons to C. elegans. Comparing the signaling systems that control vulva formation between P. pacificus and C. elegans identifies various fundamental differences. Most importantly, EGF/RAS and Wnt signaling are crucial for C. elegans vulva induction. In contrast in P. pacificus, mutations in Ppa-lin-17/Frizzled and Ppa-groucho result in gonad-independent vulva differentiation and a multivulva phenotype indicating a role in a negative signaling process. Thus, Wnt signaling has opposite roles during vulva formation in these two nematodes. We are using genetic, reverse genetic and biochemical studies to further dissect the role of Wnt signaling during P. pacificus vulva development.
To complement the macro-evolutionary comparison of two distantly related nematodes, we have initiated micro-evolutionary developmental comparisons of different Pristionchus strains and have started ecological studies. In contrast to C. elegans that lives as a soil-dwelling nematode, Pristionchus occurs in strong association with scarab beetles, in particular cockchafers and dung beetles. The results of the first year of ecological and phylogenetic analyses will be presented.
Ilan Wapinski
A genome-wide reconstruction of orthologous gene groups in fungii
Joint work with Nir Friedman, Avi Pfeffer, and Aviv Regev.
Gene duplication and divergence is a major evolutionary force. Despite the large and growing number of fully sequenced genomes, methods for identifying orthologous and paralogous relations on a genome-wide scale are still in their infancy. The most common approach is based on finding non-ambiguous, reciprocal-best BLAST hits between two genomes. While this method is simple and efficient, it often fails to disambiguate paralogous genes. Several extensions have partly addressed these problems by incorporating additional information, but none have attempted to both resolve orthologies and reconstruct their underlying evolutionary history on a genome-wide scale. Here, we propose a novel algorithm that uses sequence similarity and a given species phylogeny to resolve orthologous and paralogous relations for all genes in a large group of species simultaneously.
We applied our approach to a set of 15 fully sequenced fungal genomes spanning several hundred million years, and generated a genome wide catalog of gene trees, resolved orthologous and paralogous relations, and predicted ORF complements for all 15 species. Our reconstructed gene trees uncover which duplication or divergence events may have had the most significant effects on the sequence of genes in extant species. Focusing on the pruning of the genome following a whole genome duplication event, we can identify which duplications are preferentially maintained, and which are typically lost. Finally, by analyzing gene duplication and divergence within the context of a signaling or regulatory network, we can study how complex systems adapt to genomic changes. We postulate that gene duplications may occur with high frequency, making way for innovation due to relaxed selection on redundant copies or to the formation of back-up circuits for buffering compromised networks.
Denise Wolf
Diversity in times of adversity: probabilistic strategies in microbial
survival games
Population diversification strategies are ubiquitous among microbes, encompassing random phase-variation (RPV) of pathogenic bacteria, viral latency as observed in some bacteriophage and HIV, and the non-genetic diversity of bacterial stress responses. Precise conditions under which these diversification strategies confer an advantage have not been well defined. We develop a model of population growth conditioned on dynamical environmental and cellular states. Transitions among cellular states, in turn, may be biased by possibly noisy readings of the environment from cellular sensors. For various types of environmental dynamics and cellular sensor capability, we apply game-theoretic analysis to derive the evolutionarily stable strategy (ESS) for an organism and determine when that strategy is diversification. We find that: 1) RPV, effecting a sort of Parrondo paradox wherein random alternations between losing strategies produce a winning strategy, is selected when transitions between different selective environments cannot be sensed, 2) optimal RPV cell switching rates are a function of environmental lifecycle asymmetries and environmental autocorrelation, 3) probabilistic diversification upon entering a new environment is selected when sensors can detect environmental transitions but have poor precision in identifying new environments, 4) in the presence of excess additive noise, low- pass filtering is required for evolutionary stability, and 5) environments that select for rare phenotypes also select for diversification strategies, and define a modified Prisoner's Dilemma game. We show that even when RPV is not the ESS, it may minimize growth rate variance and the risk of extinction due to "unlucky" environment
References:
Wolf DM, Vazirani VV, Arkin AP. A microbial modified prisoner's
dilemma game: how frequency-dependent selection can lead to random
phase variation. J Theor Biol., 234(2):255-62, 2005.
Wolf DM, Vazirani VV, Arkin AP. Diversity in times of adversity:
probabilistic strategies in microbial survival games. J Theor
Biol., 234(2):227-53, 2005.
| Scientific Organizing Committee | Dannie Durand, Carnegie Mellon University, Pittsburgh, USA |
|---|---|
| Anna Tramontano, University of Rome "La Sapienza", Rome, Italy | |
| Marie-France Sagot, BAOBAB Team, INRIA Rhône-Alpes and University Claude Bernard, Lyon, France | |
| Local Organization | |
| Andrea Bandini, Elena Della Godenza, Centro Congressi di Bertinoro | |
| Sponsored by |
|
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}