|
Biological Networks III: Modularity and Genome
Evolution 3-10 June 2006 University of Bologna Residential Center Bertinoro (Forlì), Italy |
 |
|---|
|
Biological Networks III: Modularity and Genome
Evolution 3-10 June 2006 University of Bologna Residential Center Bertinoro (Forlì), Italy |
|
|---|
In 2004 and 2005, the meetings on Biological Networks I: Reconstruction, Analysis, Evolution and Biological Networks II: Interaction with Genome and Developmental Evolution brought together scientists from diverse backgrounds and perspectives for a free and exciting exchange of ideas. Both were great successes: excellent talks, good food and a relaxed atmosphere, all in a beautiful location. We have every expectation that the third meeting on Biological Networks will be just as exciting. Following the tradition established in the first two meetings, invited speakers will present new results in an environment that will promote lively, synergistic discussions of novel ideas on the computational analysis of biological networks, genome structure and dynamics, and development.
Besides the speakers, a number of PhD students are expected to participate and present their work to a small and friendly public. A call for posters will consequently be made later.
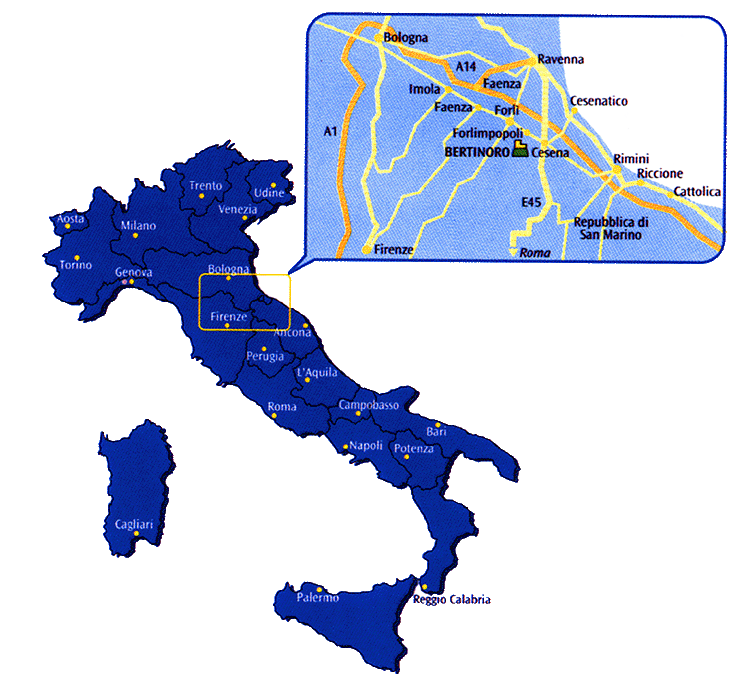
The meeting will be held in the small medieval hilltop town of Bertinoro. This town is in Emilia Romagna about 50km east of Bologna at an elevation of about 230m. Here is a map putting it in context. It is easily reached by train and taxi from Bologna and is close to many splendid Italian locations such as Ravenna and Urbino, treasure troves of byzantine art and history, and the Republic of San Marino (all within 35km) as well as some less well-known locations like the thermal springs of Fratta Terme, the Pieve di San Donato in Polenta and the castle and monastic gardens of Monte Maggio. Bertinoro can also be a base for visiting some of the better-known Italian locations such as Padua, Ferrara, Vicenza, Venice, Florence and Siena.
Bertinoro itself is picturesque, with many narrow streets and walkways winding around the central peak. The meeting will be held in a redoubtable ex-Episcopal fortress that has been converted by the University of Bologna into a modern conference center with computing facilities and Internet access.
From the fortress you can enjoy a beautiful the vista that stretches from the Tuscan Apennines to the Adriatic coast
Attention: This is still subject to some small changes.
| 08.00-09.30 | arrivals | breakfast | |||||
|---|---|---|---|---|---|---|---|
| 10.00-11.00 | arrivals | H. Margalit | J. Shapiro | C. Wiggins | J. P. Leal | departures | |
| 11.00-11.30 | coffee! | coffee! | coffee! | coffee! | |||
| 11.30-12.30 | H. Ardawatia | A. Pombo | Open problem 2 | R. Pinter | |||
| 12.30-14.30 | lunch! | lunch! | lunch! | lunch! | lunch! | lunch! | |
| 14.30-15.30 | E. Ruppin | A. Véron | M. Long | sightseeing | T. Höfer | departures | |
| 15.30-16.00 | coffee! | coffee! | coffee! | coffee! | |||
| 16.00-17.00 | T. Schlomi | G. Gonant | S. Maere | L. Mariani | |||
| 17.00-17.30 | coffee! | coffee! | coffee! | coffee! | |||
| 17.30-18.30 | Free | Free | Open problem 1 | Free | |||
Open problem sessions are meant for free discussions among the participants on a number of topics. This year, since the topics covered by the meeting are broader than last year, we wish to have more formally organized sessions. In particular:
| Arrival: | Saturday 3 June, 2006 |
|---|---|
| Departure: | Friday 9 - Saturday 10 June, 2006 |
Himanshu Ardawatia
A systematic analysis of lineage-specific evolution in metabolic
pathways
Metabolic and signaling networks have evolved over millions of years and resulted in different phenotypes. Different parts of the genome-wide metabolic networks across species evolve at different rates. Interactions in the metabolic machinery across species have profound effects on the evolution of various proteins involved in the metabolic system. Similarly the evolution of various proteins in metabolic systems alters the evolution and properties of the metabolic pathway as a whole.
We studied adaptive evolution from a systems biology perspective. In this study we investigated similarity and differences of metabolic pathway evolution across lineages and analyzed adaptive evolution in metabolic and signaling pathways across species as a function of protein evolution. As part of my talk I will present and discuss our analysis of different scenarios in lineage-specific evolution of metabolic and signaling pathways in chordates (especially human, chimp, mouse and rat).
Gavin Conant
Network evolution after genome duplication in yeast
Joint work with Ken Wolfe
A genome duplication which occurred roughly 100Mya in the lineage leading to the baker's yeast Saccharomyces cerevisiae allows us to take a somewhat unusual perspective on the study of the evolution of cellular networks. Questions raised by this event include: How was the redundancy in these networks, produced by the duplication, reduced over time to the relatively low level we see today? How do the rules which govern this loss of redundancy differ for protein-protein interaction networks as compared to gene co-expression or transcriptional regulation networks? Given the over- representation of genes involved in carbohydrate metabolism among the surviving duplicated genes, is it possible that genome duplication provided an advantage in the competition for glucose? I will discuss how various tools, including algorithms for partitioning networks, simulations of network evolution, and models of enzyme kinetics can provide insight into such questions.
Thomas Höfer
Gene regulatory networks in the immune system: mathematical
models and experiments
Abstract to be indicated later
José
B. Pereira-Leal
The contraints that protein interactions place on the functional divergence of gene duplicates (provisional title)
Abstract to be indicated later
Steven Maere
Investigating the impact of gene and genome duplications on the evolution
of development in angiosperms
Recent analysis of complete eukaryotic genome sequences has revealed that gene duplication has been rampant. Moreover, next to a continuous mode of gene duplication, in many eukaryotic organisms the complete genome has been duplicated in their evolutionary past. Such large-scale gene duplication events have been associated with important evolutionary transitions, major leaps in development, and adaptive radiations of species.M- We have developed an evolutionary model that simulates the duplication dynamics of genes, considering both genome-wide duplication events and a continuous mode of gene duplication. By applying our model to the Arabidopsis genome, for which there is compelling evidence for three whole-genome duplications, we show that gene loss is strikingly different for large-scale and small-scale duplication events and highly biased towards certain functional classes.
We provide evidence that some classes of genes were almost exclusively expanded through large-scale gene duplication events. In particular, genome duplications appear to have been crucial for the creation of a majority of important developmental and regulatory genes found in extant angiosperm plant genomes. We argue that these ancient polyploidy events might have played an important role in the origin of the angiosperms and their fast rise to ecological dominance in the Early Cretaceous, events referred to by Darwin as an 'abominable mystery'. Furthermore, we studied the expression divergence of duplicate genes and we show that the mode of duplication, the function of the genes involved, and the time since duplication are important factors in the expression divergence and therefore functional divergence of genes after duplication. By investigating the role and functional divergence of duplicate genes in the context of biological networks, we hope to be able to assess the impact of gene and genome duplications on the developmental evolution of angiosperms in more detail.
Hanah Margalit
Insights into the design and evolution of transcription regulatory networks
In my talk I will describe two studies that provide insight into the design and evolution of transcription regulation. In the first study we examined the relationship between transcription regulation and the chromosomal arrangement of transcription units (TUs). To study this relationship systematically we used network analysis methods and applied them to data from Escherichia coli and Saccharomyces cerevisiae. We demonstrate links between transcription regulation and gene adjacency and suggest that in both organisms transcription regulation has shaped the organization of TUs on the chromosome. Differences found between the organisms reflect the differences in transcription regulation between prokaryotes and eukaryotes. The second study regards the evolution of the transcription regulatory network during a relatively short evolutionary timescale. We used the well characterized transcription regulatory network of Escherichia coliK12 and followed the evolutionary changes in the repertoire of regulators and their targets across a large number of fully sequenced-proteobacteria. We demonstrate that the mode of regulation exerted by transcription factors has a strong effect on their evolution. Repressors co-evolve tightly with their target genes and are only lost from a genome once their targets have themselves been lost, or once the network has significantly rewired. In contrast, activators can be lost independently of their targets. In fact, loss of an activator can lead to efficient shutdown of an unnecessary pathway.
Luca Mariani
Stochastic gene expression in Th2 development:
a mathematical model for IL4 dynamics
In recent years, several experiments focused on the probabilistic nature of IL4 gene activation during Th2 development. We have statistically analysed the fluctuations in IL-4 protein expression, demonstrating that they are driven by high cell-intrinsic noise. This finding is in marked contrast to earlier results on bacteria and yeast, where intercellular variation is the main source of fluctuations in gene expression. The IL-4 expression data can be accounted by a multistep model of gene expression. The observed IL-4 fluctuations are reproduced by assuming gene induction to be a slow, probabilistic process. This is likely to reflect chromatin remodeling which may be restricted to a limited window of opportunity defined by T cell receptor signaling. In contrast to gene activation, the termination of expression is a rapid, deterministic process that may correlate with the loss of transcription factors from the gene (or, possibly, the recruitment of inhibitors). During the maturation of Th2 cells, both the fraction of IL-4-expressing cells and the level of IL-4 per cell increase. These two phenomena can be accounted for by more rapid chromatin opening (e.g., through a more accessible basal chromatin state) and higher efficiency of transcription in mature Th2 memory cells.
Ron Pinter
Faithful modeling of transient behavior in developmental
pathways
Joint work with Amir Rubinstein, Vyacheslav Gurevich, and Yona Kassir.
The modeling and analysis of genetic regulatory networks is essential both for better understanding their behavior as well as for elucidating and refining open issues. Many methods for simulating and inspecting the properties of such pathways have been devised, borrowing from a variety of techniques such as differential equations, algebraic calculi, and Flux Balance Analysis. Most of these methods are quantitative in nature and require data that is often not fully revealed; moreover, some of them are computationally intensive, requiring significant time and resource.
We present a computational model that allows for qualitative analysis of regulatory pathways, enabling the examination of characteristics such as transient behavior, robustness, and sensitivity to initial conditions, in an effective manner. To this end, we have extended the Boolean network model, which has limited modeling power, to a richer albeit discrete network model, while maintaining computational efficiency. Moreover, we have borrowed a simple technique for the representation of functions, namely Karnaugh maps, to elucidate and visualize the behavior of the pathways under study. We have applied our method to analyze the transience and robustness of a representative developmental pathway, namely early meiosis in budding yeast. Some of our analytic observations, such as the pathway's response to premature expression of a key regulator, were validated in the lab and were found to be in agreement with experimental data. Furthermore, our analysis predicts new modes of regulation by which negative feedback loops accomplish their roles.
Ana Pombo
Intermingling of chromosome territories, translocations and
interchromosomal associations
During interphase chromosomes occupy distinct territories in the mammalian cell nucleus. Current models suggest that there is little or no intermingling between individual chromosome territories (CTs), and propose the existence of an interchromatin domain (ICD) compartment where only rare inter-chromosomal interactions can occur via extended chromatin loops. This type of organization is difficult to reconcile with measurements of chromatin dynamics and frequency of chromosomal translocations, which suggest a higher extent of interchromosomal interactions. Using an improved fluorescence in situ hybridization procedure on thin (~150 nm) cryosections (cryo-FISH), we show that CTs intermingle to a significant extent, with ~20% of the genome present in intermingled conformations in activated lymphocytes. Intermingling is also seen at nanometer resolution, on the electron microscope, where DNA fibers from different chromosomes lie in cell nucleus. Current models suggest that there is little or no intermingling between individual chromosome territories (CTs), and propose the existence of an interchromatin domain (ICD) compartment where only rare inter-chromosomal interactions can occur via extended chromatin loops. This type of organization is difficult to reconcile with measurements of chromatin dynamics and frequency of chromosomal translocations, which suggest a higher extent of interchromosomal interactions. Using an improved fluorescence in situ hybridization procedure on thin (~150 nm) cryosections (cryo-FISH), we show that CTs intermingle to a significant extent, with ~20% of the genome present in intermingled conformations in activated lymphocytes. Intermingling is also seen at nanometer resolution, on the electron microscope, where DNA fibers from different chromosomes lie in close proximity, creating the potential for functional interactions. We find that the extent of intermingling between chromosome pairs in human lymphocytes correlates with the frequency of chromosome translocations in the same cell type. Blocking of transcription revealed the existence of transcription-dependent interchromosomal interactions that influence chromosome organization in the nucleus. This is further supported by the detection of the active form of RNA polymerase II in areas of intermingling. Thus, chromosome organization in the nucleus seems to depend on inter-chromosomal interactions that occur during gene transcription and that can lead to cell-type specific chromosome arrangements.
Eytan Ruppin
Genetic Robustness and Annotation: A System's View of Metabolism.
Joint work with David Deutscher, Martin Kupiec and Isaac Meilijson (Genetic robustness) and Tomer Shlomi, Efrat Naim, Markus Herrgard and Roded Sharan (Genetic Annotation).
In this talk I shall review two recent studies in my lab, utilizing constraint-based metabolic models (CBM) to investigate two fundamental concepts: Genetic robustness and Genetic annotation. To study genetic robustness, i.e., the constancy of the phenotype in face of heritable perturbations, we conduct an in-silico multiple-knockout investigation of a CBM model of the yeast's metabolic network. We find that 74% (360) of the metabolic genes participate in processes that are essential to growth already in a standard laboratory environment, compared with only 13% previously found to be essential using single knockouts. Alternative pathways comprise a more dominant mechanism than duplication, underlying 45-78% of backed-up genes. The genes' interaction depth is shown to be a solid indicator of their biological buffering capacity. It is correlated with both the genes' environmental specificity and their evolutionary retention. These results provide the first multiple knockout account of robustness in a large scale biological network. To study gene annotation, we use CBM models to investigate the functioning of genes under multiple environmental and genetic conditions. We derive a high-dimensional annotation (HD) of metabolic genes, which associates genes with terms representing metabolic processes under various contexts. These contexts are spanned by three dimensions representing growth media, the availability of oxygen, and genetic environment. Comparing the obtained HD-annotation with GO provides a set of novel annotations. These have been tested and validated computationally, using gene conservation and expression data. They are now also being tested experimentally, by conducting corresponding growth experiments in the pertaining contexts.
James Shapiro
Basic considerations in formulating a 21st Century theory of the
genome and genome evolution.
Molecular studies, going back to Monod's pioneering work on glucose/lactose metabolism, have shown that cells are sophisticated cognitive and computing entities. The interactive and allosteric properties of biological macromolecules facilitate the formation of signal transduction networks linking sensory and information-processing molecules distributed throughout the cell. How do various genome functions fit into cellular informatic networks? Classical genetic concepts of genotype and phenotype developed from a mechanical, linear view of cell function and cybernetics and cannot answer this question adequately. Classical concepts of unidirectional information transfer are incompatible with our molecular understanding that cellular DNA only functions in nucleoprotein complexes. Starting with a more contemporary model of the genome as a highly formatted information storage organelle, we can develop more satisfactory concepts of genome organization and function. I will explore how the concepts of natural genetic engineering and genome system architecture provide new ways of thinking about DNA reorganization in evolution.
Shlomi Tomer
Conservation of Expression and Sequence of Metabolic Genes is Reflected by Activity Across Metabolic States.
Joint work with Yonatan Bilu, Naama Barkai and Eytan Ruppin.
Variation in gene expression levels on a genomic-scale has been detected among different strains, among closely related species and within populations of genetically identical cells. What are the driving forces which lead to expression divergence in some genes, and conserved expression in others? Here we employ Flux Balance Analysis (FBA) to address this question for metabolic genes. We consider the genome-scale metabolic model of S. cerevisiae, and its entire space of optimal and near-optimal flux distributions. We show that this space reveals underlying evolutionary constraints on expression regulation, as well as on the conservation of the underlying gene sequences. Genes which have a high range of optimal flux levels tend to display divergent expression levels among different yeast strains and species. This suggests that gene regulation has diverged in those parts of the metabolic network which are less constrained. In addition, we show that genes which are active in a large fraction of the space of optimal solutions tend to have conserved sequences. This supports the possibility that there is less selective pressure to maintain genes which are relevant for only a small number of metabolic states.
Amélie Véron
Evolution of protein-protein interaction networks
Joint work with Kerstin Kaufmann and Erich Bornberg-Bauer
As organism complexity correlates with an increase of both the absolute number of transcription factors (TF) and their proportion in the genome, the study and reconstruction of the evolution of regulatory networks is essential to understand the formation of new species, the development of organisms, their evolutionary and physiological adaptation to environmental changes and suggests points of intervention for genetic manipulations.
The protein-protein interaction (PPI) networks formed by TF lie at the heart of regulatory systems, as different TF dimers regulate the expression of different target genes in different ways and different tissues. To date, a vast number of biological networks and especially PPI networks have been shown to display hub-based topologies, i.e. some proteins having many interactions and many having few and, closely related to this, the characteristic property of so called scale-free-ness.
In the recent past, our study of such TF PPI networks in metazoans -- the bHLH, bZIP and NR PPI networks -- led us to the conclusion that, based on the standard genetic evolutionary mechanisms, different network topologies could emerge. In particular, the relative importance of single gene duplication versus whole genome duplication events and the gain and loss of protein domains seem to be two crucial factors shaping PPI networks.
In plants, polyploidy and thus whole genome duplications are far more frequent than in metazoan genomes. Therefore, the study of a plant TF PPI network could help understanding the influence of whole genome duplications on the evolution of a network. The MADS-domain proteins, and especially the MIKC-like proteins, which we studied, play an important role in both the vegetative and the reproductive phases of plant life, and are thus an intensively studied transcription factor family. By combining phylogeny, domain analysis and PPI data from different species, we show that the MADS-domain MIKC-type PPI network displays unique properties that can be explained by an evolutionary model of whole genome duplications followed by loss of interactions.
| Scientific Organizing Committee | Dannie Durand, Carnegie Mellon University, Pittsburgh, USA |
|---|---|
| Anna Tramontano, University of Rome "La Sapienza", Rome, Italy | |
| Marie-France Sagot, BAOBAB Team, INRIA Rhône-Alpes and University Claude Bernard, Lyon, France | |
| Local Organization | |
| Andrea Bandini, Eleonora Campori, Centro Congressi di Bertinoro | |
| Sponsored by |
|
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}