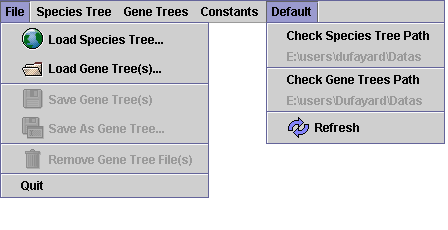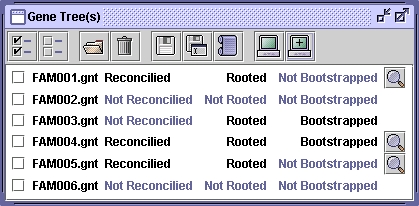
From this point, you can load, root, reconcile and save gene trees. You can numerate orthologous gene families and save results in a text file.
 Select every tree of the list.
Select every tree of the list.
 Unselect every tree of the list.
Unselect every tree of the list.
 Add gene tree file(s). They consists in text files containing each gene with the corresponding species, and a newick gene tree with branch lengths and eventually a "bootstrap" value. These trees can be found at: http://pbil.univ-lyon1.fr/databases/hovergen.html For example:
Add gene tree file(s). They consists in text files containing each gene with the corresponding species, and a newick gene tree with branch lengths and eventually a "bootstrap" value. These trees can be found at: http://pbil.univ-lyon1.fr/databases/hovergen.html For example:
[
OAAIII.PE1 Ovis aries
CHKSPINH.PE1 Gallus gallus
HUMATH3U7.AT3 Homo sapiens
HUMATH3A2.AT3 Homo sapiens
S47225.PE1 Mus sp.
S79838 Gallus gallus
AF006495.PE1 Cyprinus carpio
AB027238.PE1 Cavia porcellus
AB026832.PE1 Takifugu rubripes
ASA252153.PE1 Salmo salar
]
(OAAIII.PE1:0.07650,(HUMATH3U7.AT3:0.03050,HUMATH3A2.AT3:0.00000):0.01600,(S47225.PE1:0.08000,(AB027238.PE1:0.11550,((CHKSPINH.PE1:0.30000,S79838:0.02900):0.11750,(AB026832.PE1:0.14850,(AF006495.PE1:0.17000,ASA252153.PE1:0.13300):0.01350):0.07400):0.07200):0.00750):0.00050);
 Remove from the list any selected gene tree file.
Remove from the list any selected gene tree file.
 Save in the same files selected gene trees with current root and reconciliation.
Save in the same files selected gene trees with current root and reconciliation.
 Save in another file the first selected gene tree with current root and reconciliation.
Save in another file the first selected gene tree with current root and reconciliation.
 Save in a text file orthologous gene families issued from every selected file. For example:
Save in a text file orthologous gene families issued from every selected file. For example:
FAM001.gnt 1 AF006495.PE1
FAM001.gnt 1 AB026832.PE1
FAM001.gnt 1 ASA252153.PE1
FAM001.gnt 1 HUMATH3A2.AT3
FAM001.gnt 1 HUMATH3U7.AT3
FAM001.gnt 1 AB027238.PE1
FAM001.gnt 1 S47225.PE1
FAM001.gnt 1 OAAIII.PE1
FAM001.gnt 1.1 CHKSPINH.PE1
FAM001.gnt 1.2 S79838
 Reconcile every selected and rooted gene tree. An exhaustive species tree must be loaded.
Reconcile every selected and rooted gene tree. An exhaustive species tree must be loaded.
 Reconcile and choose a root for every selected gene tree. An exhaustive species tree must be loaded.
Reconcile and choose a root for every selected gene tree. An exhaustive species tree must be loaded.
 Open the results window of the corresponding tree.
Open the results window of the corresponding tree.