


Principle of RFDD
Restriction Fragment Differential Display is a cDNA AFLP-derived technique dedicated to transcriptomic analysis. It will provide you qualitative determination of variations between two samples as small as 1-2 micrograms total RNA. Potentially, all transcripts can be detected as long as they are known in the RefSeq database from NCBI and have restriction sites for the chosen enzymes.
1- At the bench
1-1- Extract total RNAs from different samples.
For each sample, follow steps 1-2 to 1-7.
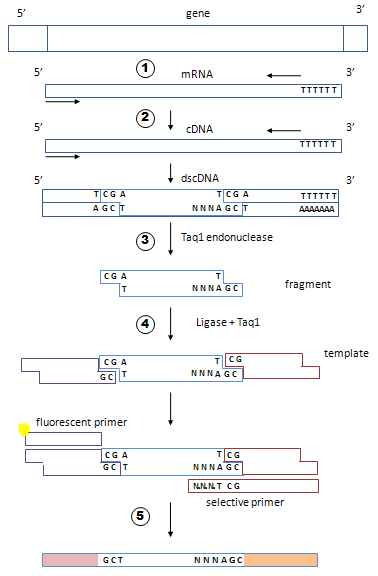
1-2- Prepare double-stranded cDNAs. Reverse transcription may be done with an oligo dT priming or with a random primer mix.
1-3- Fragment your cDNAs with a restriction enzyme, for example TaqI endonuclease that recognizes the sequence TCGA and cleaves between T and C.
1-4- Make a ligation with a mix of 2 adapters, A1 and A2, in the presence of TaqI to prevent ligation between fragments. You obtain a mix of adapted fragments.
1-5- Make 64 PCRs. For this use two types of primers. The fluorescent primer will hybridize adapter A1. The selective primer will hybridize adapter A2 plus the remaining restriction site plus 3 nucleotides flanking the restriction site. Since there are 43 combinations of 3 nucleotides, there are 64 selective primers. In each of the 64 tubes, prime the PCR with the fluorescent primer associated with one of the 64 discriminating primers.
If the same triplet is present on both ends of the same fragment (rare event) the fragment adapted to 2 adapters A2 can be amplified, but will not be fluorescent. If the ends are different, fragments adapted to 2 adapters A2 cannot be amplified. Similarly, fragments adapted to 2 adapters A1 cannot be amplified because by design A1 antisense strand cannot be elongated. At the end you obtain mainly fluorescent amplicons originating from the fragments adapted to A1 and A2 (in both combinations).
1-6- Separate the PCR products by electrophoresis on a capillary sequencer in order to obtain a good resolution.
1-7- You get 64 lists of amplicons. For each amplicon, you know the size and 11 nucleotides of the original RNA: 8 nucleotides of the two restriction sites plus 3 selective nucleotides flanking one of the restriction site. Each restriction fragment should be amplified by two different PCRs discriminating its two different ends.
Ideally, you could identify each amplicon without any further experimental procedure, provided that a database of all possible amplicons has been built. It is the aim of this website to organize your data and to associate a candidate transcript to each amplicon.
In fact, there can be several candidates for an amplicon. However, since a transcript can be detected by several PCRs, this website crosschecks your results in order to eliminate false positives (see step 2-3-). This crosschecking will be much better if you use 2 or more different restriction enzymes to fragment your cDNA in duplicate experiments, and, so doing, if you make 128 PCRs per sample instead of 64.
2- At the computer
Use this website to organize your data and identify the transcripts from their amplicons and from the RFDB database (RFDB can also be accessed independly).
2-1- Organize your results. Enter the files containing the list of all amplicons found (characterized by their selective triplet, size and fluorescence level). The software will align the matching amplicons from 2 different experimental conditions. The output is a raw data file of paired amplicons that you can save (see 2-4).

2-2- Identify the candidate transcripts corresponding to your amplicons. The website connects your data to a database of predicted amplicons (RFDB database), and therefore provides a new list of your amplicons, their variations between 2 experimental conditions and one or several candidate transcripts for each amplicon.

2-3- If there are several candidate transcripts per amplicon, how to decide between? By crosschecking your results. A fragmented transcript can be detected by two amplicons (adapter A2 can alternatively bind the two ends of one fragment), or more (there can be several fragments). To cross check, the software will sort your amplicon list by candidate-transcript, then discard those detected by only one amplicon. When several amplicons are found for one candidate transcript, a majority must vary in the same way (increased in condition 2, for instance). If no majority is found, the candidate is discarded. After this in silico validation, you obtain a final list of transcripts with their variation between two experimental conditions.
2-4- Optional. Your data from step 2-1 can be re-analyzed later in the future when the genome annotations will have improved, and then get new results.