Database Building
Sequences
The database is built using sequences taken from SWISS-PROT and its annex
TrEMBL. The reasons that led us to choose these collections are threefold:
- The SWISS-PROT + TrEMBL set is exhaustive and non-redundant.
- The annotations are of high quality compared to the other general
sequence database systems.
- Almost all entries are cross-referenced with their corresponding
nucleotide sequences in GenBank / EMBL / DDBJ.
Duplication
In SWISS-PROT+TrEMBL, homologous
genes from different organisms (or paralogous genes within a genome) that code for a same protein are described by a single entry.
In the view of HOINVGEN, there should be a specific entry for each gene to allow one to retrieve all existing homologous genes.
For these reasons the SWISS-PROT + TrEMBL entries that correspond to several genes are duplicated before being used for families
calculation. These duplicated entries are
given a new name. The duplicated sequences are signaled by their
annotations in the CC field:
Example: The sequence A70A_DROSI presents the following annotation:
CC -!- modified from A70A_DROMA.
Families
To build the families we perform a similarity search of all the proteins
against each other with
BLASTP2.
For this purpose, we use the BLOSUM62 similarity matrix and a threshold of
10-4 for E-values. Low complexity sequences
are filtered with SEG.
Then, the results are processed this way:
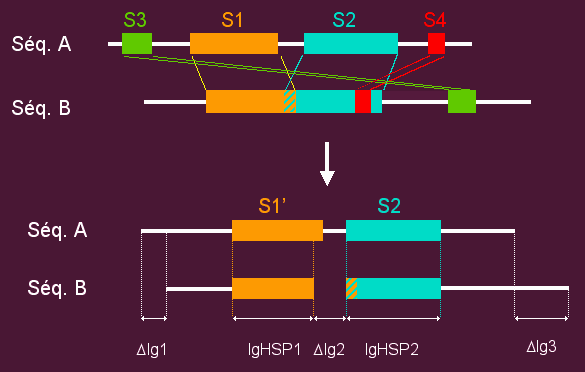
- For each pair of sequences, Homologous Segment Pairs (HSPs) that are not
compatible with a global alignment are removed (see example).
- Two sequences in a pair are included in the same family if:
- The remaining HSPs cover at least 80% of the proteins length.
- Their similarity is greater or equal to 50% (two amino-acids are
considered similar if the BLOSUM62 similarity score is positive).
- Both sequences are complete.
- We use simple transitive links to build our families. If a pair of
sequences named A + B and a pair of sequences B + C fulfill the conditions
listed above, then A, B and C are integrated in the same family, this even if
the pair A + C does not fulfill these conditions.
- Once families of complete protein sequences have been build, partial
sequences (longer than 100 AA or at least 50% of the length of the complete
proteins) are included in the classification. A partial sequence matching
with a complete protein is included in its family if:
- The remaining HSPs cover at least 80% of the partial protein length.
- Their similarity is greater or equal to 50%.
- Short partial sequences (less than 100 AA and less than 50% of the length of
the complete proteins) are not included in the classification.
Alignments
For each family, protein sequences are aligned using
CLUSTALW 1.83. All
the default parameters are used excepted that the "Fast/Approximate" option is
preferred for pairwise alignments.
Phylogenetic trees
The phylogenetic trees are computed with
TREE-PUZZLE 5.2 by maximum likelihood. Currently only trees
with a minimum of 4 and a maximum of 257 (3,243 out of 10,073) protein
sequences are reconstructed.
If you have problems or comments...
 Back to PBIL home page
Back to PBIL home page
{kind=link}