Atelier INSERM: Bioinformatique
Partie Phylogénie
11 Mai 2001
Le TP utilisera principalement deux programmes
Un editeur d'alignement multiple de sequences seaview
Un programme de phylogenie avec parcimonie et NJ : phylo_win
Ces programmes sont utilisables sur votre station de travail.
A) Mise en jambes : la phylogénie universelle
Le fichier 28sfrags.mase contient un ensemble de sequences d'ARNr de la grande sous-unité (LSU) d'eucaryotes et procaryotes pré-alignées.
Visionner l'ensemble de sites "bien" alignes nommé "all sequences"
Construire la phylogenie universelle
La bootstrapper
Essayer la distance Transversions-seules
La position de la sequence chloroplastique d'Euglena est-elle attendue?
B) Une bacterie vieille de 250 MA : est-ce possible ?
Vreeland et al. ont publie avoir isole une bacterie vieille de 250 MA
dans un cristal de sel.
Leurs donnees sont reproduites dans le fichier de sequences d'ARNr 16S bacteriens alignes permians.mase. Que pensez-vous de leurs conclusions? Comparer les approches par parcimonie et par distances + NJ.
C) L'origine de HIV-1 et HIV-2 parmi les virus de primates SIV
Gao et al. ont publie (Nature 397:436) une analyse phylogenetique du gene pol des virus HIV-1 et HIV-2 et de leurs homologues simiens. Le fichier hivpol.mase rassemble les donnees publiques de sequences proteiques sur lesquelles tenter de reproduire leurs resultats. La sequence FIV/Oma (Feline Immunideficiency Virus) sert de groupe externe a l'analyse. Le fichier hivpol-dna.mase reproduit le meme alignement au niveau nucleotidique.
Identifier quelles especes de singes sont a l'origine de HIV-1 et HIV-2?
Mener des analyses sur distances Ka puis sur distances Ks dans les cas ou c'est possible.
Le fichier hivpol.pdf contient l'article (presque) complet de Gao et al. (il y a un probleme avec sa 1ere page!).
D) Tester l'hypothese OPV : Oral Polio Vaccine
L'hypothese que le virus HIV-1 aurait ete transmis a l'homme par des preparations vaccinales contre la polio utilisees en Afrique dans les annees 50 a ete formulee. Les donnees du fichier frag12s.mase sont les donnees brutes de l'article de Blancou et al. qui refute cette hypothese.
Ce sont des sequences non alignees d'un fragment de ~140 pb du gene de l'ARNr 12S mitochondrial. Il faudrait leur rajouter les sequences homologues des genres Homo, Pan, Gorilla, Cercopithecus, Erythrocebus, Cercocebus, Macaca, les aligner, et reproduire l'analyse de Blancou et al.
Berry et al. ont independemment publie une refutation de la meme hypothese OPV. Malheureusement leurs donnees ne semblent pas disponibles.
En cas de difficulte a trouver les sequences, le fichier 12sprimates.mase donne du materiel.
E) Une situation avec l'artefact "Long Branch Attraction"
Le fichier microsplsu.mase contient l'alignement de plusieurs sequences d'ARNr de la grande sous-unite (LSU) de divers eucaryotes et archaea. Il contient aussi le LSU-ARNr de la microsporidie Encephalitozoon cuniculi, groupe de protistes dont la position phylogenetique est tres debattue.
Constater combien la sequence de microsporidie est reduite par rapport aux autres sequences eucaryotes
Remarquer que les LSU rRNA ne sont pas alignables sur toute leur longueur entre eucaryotes distants mais seulement sur certaines parties mieux conservees. Essayer de selectionner un ensemble de sites "bien alignes".
Quelle origine evolutive est predite pour les microsporidies par ces sequences etudiees avec une distance comme K2P et NJ ?
Neanmoins, Peyretaillade et al. ont publie (NAR 26:3513) une analyse de ces donnees avec et sans prise en compte de la variabilite de la vitesse d'evolution des differentes parties de la molecule. Constater la difference entre ces deux inferences phylogenetiques sur les memes donnees.
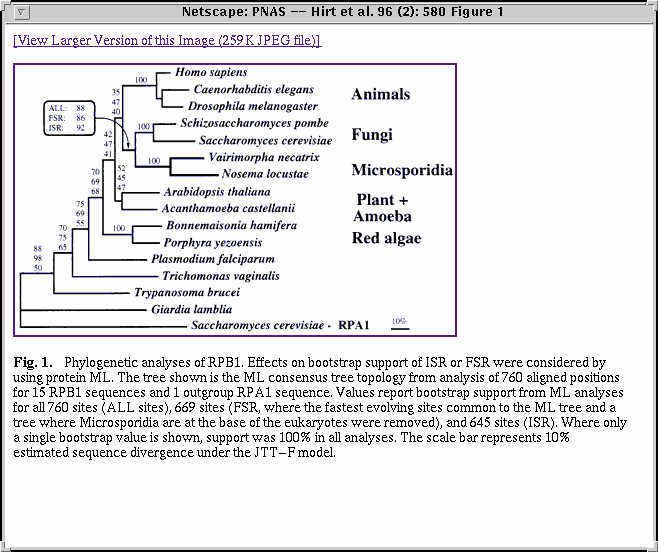
Le fichier rpb1.mase contient l'alignement de plusieurs sequences de la proteine RPB1 (grande sous-unite de la RNA polymerase II) de divers eucaryotes et de deux microsporidies, Vairimorpha necatrix et Nosema locustae. Les sequences RPC1_YEAST et RPA1_Yeast forment un groupe externe.
Hirt et al. (PNAS 96:580) ont analyse ces donnees au maximum de vraisemblance (programme ProtML).
Analyser ces donnees par distances proteiques + NJ + bootstrap.
Etudier les choix de sites "bien" alignes
Visionner les arbres (precalcules) sur ces donnees (choix de sites choix2) par ProtML avec et sans groupe-externe.
Comparer la position des lignees sur des longues branches.
{kind=link}