|
|
|
Guy Perrière
Laboratoire de Biométrie et Biologie Évolutive
UMR CNRS 5558
Ce TP a pour objet l'étude comparative de l'usage des codons chez deux bactéries fort différentes : Escherichia coli K12 et Neisseria meningitidis Z2491 (sérogroupe A). E. coli est une entérobactérie fort répandue, et la souche K12 est celle que l'on trouve au niveau de l'intestin des mammifères. N. meningitidis est une bactérie pathogène responsable de méningites. Afin d'étudier l'usage du code génétique chez ces deux organismes, nous allons utiliser un ensemble de mesures statistiques que nous manipulerons au moyen du logiciel Excel.
Parmi ces mesures statistiques, figure une Analyse Factorielle des Correspondances (AFC) sur la composition en codons des gènes. Cette méthode permet de représenter sur un plan (graphique à deux dimensions) les proximités entre individus décrits par un grand nombre de variables. Une AFC produit ce que l'on appelle des facteurs, est c'est le croisement de ces facteurs qui permet de construire les plans en question (on parle de plans factoriels). Le nombre de facteurs produits par une AFC est égal au nombre de variables – 1, c'est-à-dire 63 dans le cas d'une analyse réalisée sur un tableau de fréquence des codons. Généralement, seuls les trois ou quatre premiers facteurs sont significatifs, et peuvent être utilisés pour construire des plans factoriels. Les facteurs sont numérotés par ordre de significativité décroissante. Cela signifie que les proximités entre les individus rapportées par le premier facteur correspondent à une tendance plus forte que celle rapportées par le deuxième, elle-même plus importantes que celle observée sur le troisième, etc.
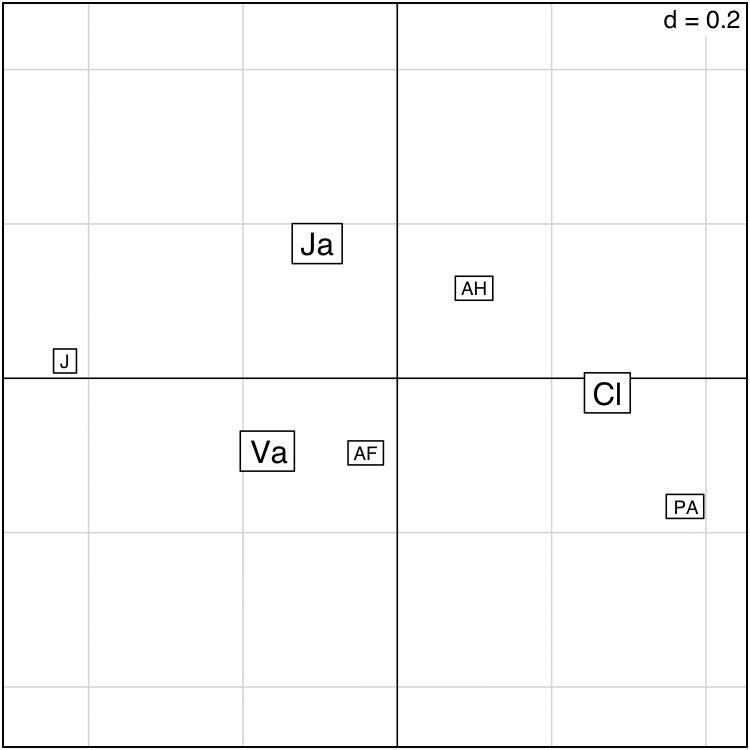
Un exemple simple d'AFC figure ci-dessous. Dans le tableau de départ, quatre catégories d'individus (Jeunes = J, AF = Adulte Femme, AH = Adulte Homme, PA = Personnes Agées) sont décrits par trois variables correspondant au style de musique écoutée (Variétés = Va, Jazz = Ja, Classique = Cl). L'AFC calculée sur ce tableau va produire deux facteurs, et le croisement de ces deux facteurs donne le graphique à droite :
|
|
|
Sur ce plan factoriel on visualise directement l'échelonnement des goûts musicaux en fonction des individus. Les jeunes ont des gouts à l'opposé de ceux des personnes âgées, tandis que les adultes hommes et femmes sont plus proches. Les hommes adultes sont partagés entre jazz et musique classique tandis que les femmes adultes ont plus tendance à écouter de la variété.
L'analyse effectuée chez E. coli porte sur l'ensemble des 4254 gènes de cet organisme dont la longueur est >150 nucléotides. En effet, on considère que des gènes courts peuvent avoir une composition en codons biaisée du fait fait de leur taille.
Dans un premier temps, nous allons récupérer les données sur ces gènes. Pour ce faire, cliquez sur le lien ci-dessous :
http://pbil.univ-lyon1.fr/members/perriere/cours/MSBM/Ecoli.txt
Sauvez les données au format Texte , ceci en utilisant
l'option Enregistrez sous d'Internet Explorer. Comme nom de
fichier, utilisez celui qui est donné par défaut.
Après lancement du programme, utilisez l'option Ouvrir du menu
Fichier pour charger le fichier Ecoli.txt. Dans la
boîte de dialogue qui apparait, cliquez tout de suite sur le bouton
Terminer. Il se peut que des lignes vides se trouvent en début de
fichier (bug dû à Internet Explorer). Dans ce cas, sélectionnez les lignes
en question et supprimez-les à l'aide de l'option Supprimer du menu
Edition. Vérifiez que votre fichier contient bien 4255 lignes.
La signification des différentes colonnes du fichier est la suivante :
Pour interpréter les résultats, il est nécessaire de tracer quelques graphes. Pour commencer, nous allons visualiser le nuage de points croisant les deux premiers facteurs de l'AFC. Sélectionnez la la seconde et la troisième colonne, puis cliquez sur le bouton de l'assistant graphique :
Sélectionnez l'option Nuage de points puis cliquez sur le bouton
Suivant en gardant toutes les options par défaut jusqu'à arriver à
l'étape de sélection de l'emplacement du graphique. A ce niveau, sélectionnez
l'option sur une nouvelle feuille. Cette option crée un onglet sur
le classeur qui permet de passer facilement du tableau de données au graphe,
nous l'utiliserons pour tous les graphes construits au cours de ces TP :
Le nuage de points présente une structuration assez forte, ce qui montre qu'il
existe bien des tendances particulières au niveau de la composition en codons
pour les gènes d'E. coli. Réitérez l'opération de construction de
graphe pour les facteurs F1 et F3 de l'analyse. Remarque : pour
sélectionner deux colonnes non adjacentes, maintenez la touche Alt
Gr enfoncée tout en cliquant sur la deuxième colonne. Que constatez-vous
concernant le facteur F3 ?
Maintenant, construisez un nuage de points croisant les valeurs de F1 avec celles
de 1/CAI. Une fois le graphe obtenu, nous allons ajouter une courbe de tendance
pour mesurer le coefficient de corrélation entre ces deux valeurs. Pour ce faire,
positionnez-vous avec la souris au centre du nuage de points puis faites
apparaître le menu contextuel d'Excel en cliquant sur le bouton droit de
la souris, ensuite sélectionnez l'option Ajouter une courbe de
tendance. Dans l'onglet Type, sélectionnez
Linéaire :

Puis, dans l'onglet Options, cochez la case Afficher le
coefficient de détermination sur le graphique :
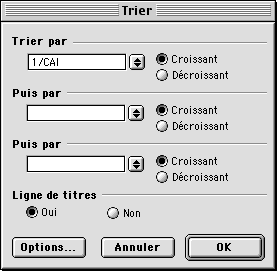
La droite de régression se superpose alors au nuage de points, avec affichage de la valeur de R2. Comme nous l'avons vu dans le cours, cela montre bien que le biais principal qui affecte la composition en codons chez E. coli est l'expressivité des gènes puisque le facteur principal de l'AFC est corrélé de façon significative à l'inverse du CAI. Pour confirmer cette observation, il est possible de trier le classeur en fonction des valeurs décroissantes de CAI et de regarder les noms des séquences possédant les plus hautes valeurs.
Pour trier le classeur, cliquez tout d'abord sur la zone grise située sur le
bord supérieur gauche du tableau. Ceci a pour effet de sélectionner
l'intégralité des lignes et des colonnes. Ensuite, sélectionnez l'option
Trier qui figure dans le menu Données. Choisissez de
trier par valeurs croissantes de 1/CAI :
Le code à cinq caractères figurant après le nom de la séquence
(ECOLICG) correspond au nom du gène. Vous devez constater que
beaucoup de séquences dont le nom est de la forme ECOLICG.RP** figurent
maintenant dans les 50 premières lignes du tableau. Les dites séquences codent
pour des protéines ribosomiques dont on sait qu'elles sont fortement exprimées
chez les bactéries. Les gènes lpp ou omp* correspondent à des
protéines majeures de la membrane plasmique, eno code pour l'énolase, une
enzyme clé de la glycolyse, tufA, tufB, tsf et tig
pour des protéines intervenant dans l'élongation peptidique. Tous ces gènes sont
fortement exprimés chez la plupart des bactéries cultivables.
Réitérez l'opération de construction de graphique avec affichage de la courbe de tendance accompagnée de son coefficient de détermination pour le nuage de points croisant les colonnes F2 et G+C%, puis pour le nuage de points croisant les colonnes F3 et KD. Vous devez constater que les dits facteurs sont effectivement corrélés de façon significative avec ces indices chez E. coli. Qu'en déduisez-vous quant aux facteurs principaux gouvernant l'usage du code chez E. coli. ?
Maintenant que vous maîtrisez l'utilisation d'Excel, nous allons passer à un exercice d'application complet sur un autre génome. Vous allez donc réitérer toutes les analyses effectuées précédemment sur les 2121 gènes (de longueur >150 nucléotides) de N. meningitidis. Le tableau contenant les valeurs des différents indices et mesures statistiques est disponible ci-dessous :
http://pbil.univ-lyon1.fr/members/perriere/cours/MSBM/Nm.txt
Notez qu'un indice supplémentaire a été calculé dans ce cas : G+C3%, il s'agit simplement du pourcentage de bases G+C en troisième position des codons. En effet, du fait de la dégénerescence du code génétique, une bonne partie de la plasticité de la composition en bases va jouer sur cette position.
Une fois que vous avez importé toutes les données sous Excel, regardez avec quel indice le premier facteur de l'AFC est le plus fortement corrélé. Est-ce avec l'inverse du CAI (1/CAI) ? Ou autre chose ? Essayez les différents couples facteur/indice pour voir qu'est-ce qui est le plus fortement corrélé avec quoi. Poussez l'analyse jusqu'au quatrième facteur de l'AFC. Y a-t-il des différences importantes avec E. coli ?
Maintenant, triez votre classeur Excel par valeurs croissantes de G+C3%.
Sélectionnez alors les 60 premiers noms de séquences et copiez-les dans le
tampon, en utilisant l'option Copier du menu Edition.
Cliquez sur le lien ci-dessous, qui vous permet de définir une liste de
séquences sur le serveur Web du PBIL (Pôle Bioinformatique Lyonnais) :
http://pbil.univ-lyon1.fr/search/list.php
Une fois la fenêtre ouverte, coller la liste de noms dans le champ
Paste your list below. Sélectionnez la banque EMGLib à
l'aide du menu déroulant situe à côté du champ List name. Une
fois ceci fait, cliquez sur le bouton Submit.
Dès que la page Sending output apparaît, il est possible
d'accéder aux séquences correspondant aux noms que nous avons envoyés. Pour ce
faire, il suffit simplement de cliquer sur leur nom (e.g.
NEIMACG.NMA0023). Pour que l'intégralité des séquences figurent
dans la page, vous pouvez selectionner 100 dans l'option
Display per page.
Explorez maintenant les annotations qui sont associées à chacun de ces gènes.
Intéressez-vous plus particulierement au champ /product qui donne
le nom du produit (i.e. la protéine) associée au gène. Que remarquez-vous
à ce niveau ? Quelles est la catégorie fonctionnelle la plus fréquemment
rencontrée ?
Nous allons maintenant nous intéresser à un cas particulier : le gène
NMA1500. Vous allez chercher quels sont les plus proches homologues de ce
gène dans d'autres espèces que N. meningitidis. Pour ce faire, cliquez
tout d'abord sur le lien vous permettant d'accéder à la séquence
(NEIMACG.NMA1500). Une fois la page chargée, sélectionnez à la
souris la séquence protéique (disponible au niveau du champ /translation)
et copiez-là dans le tampon. Une fois ceci fait, cliquez sur le lien ci-dessous pour
accéder au système de recherche d'homologues du NCBI :
http://www.ncbi.nlm.nih.gov/blast/
Dans la fenêtre Enter accession number, gi, or FASTA sequence,
collez la séquence protéique du gène nma1500. Lancez ensuite le programme de
recherche au moyen du bouton BLAST.
Dans la fenêtre de résultats apparaissent alors les séquences
protéiques les plus similaires à la séquence envoyée comme requête.
L'ordre dans lequel les séquences sont rangées est celui de la similarité
décroissante. Encore une fois, en cliquant sur les noms des séquences
vous pouvez accéder à celles-ci. En examinant les annotations des séquences,
determinez quelles sont les espèces pour lesquelles la similarité détectée avec
le gène nma1500 de N. meningitidis est la plus forte (champ
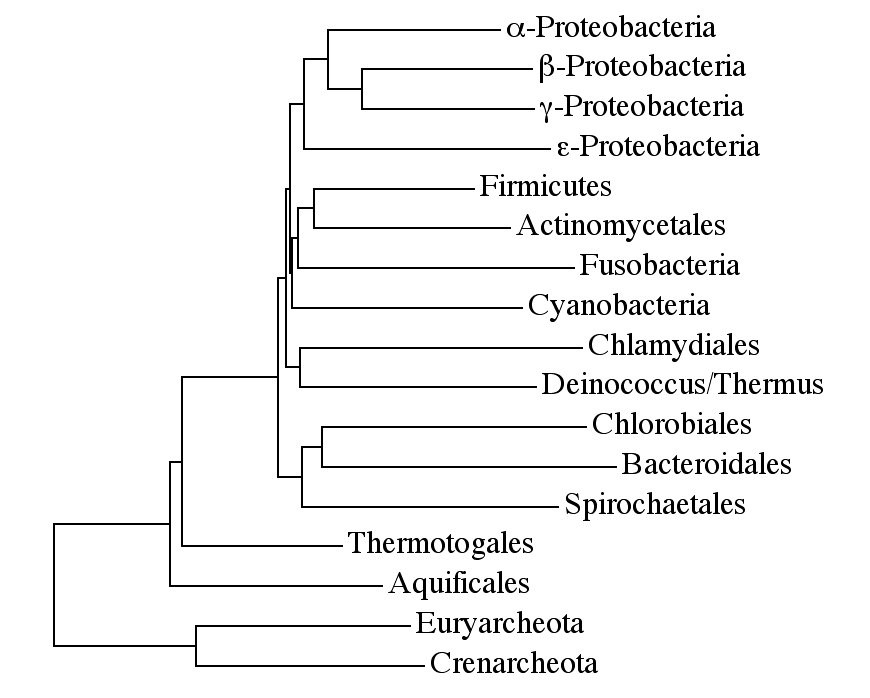
ORGANISM des annotations). Sachant que N. meningitidis est
une β-Protéobacterie et, en vous aidant de l'arbre phylogénétique
disponible ci-dessous que pouvez-vous conclure quant au gène nma1500 ?
 Back to PBIL home page
Back to PBIL home page