|
|

|
|
ChromoNet |
Purpose
A classical image used by computational biologists when speaking about gene regulation is
the one represented in the figure below.
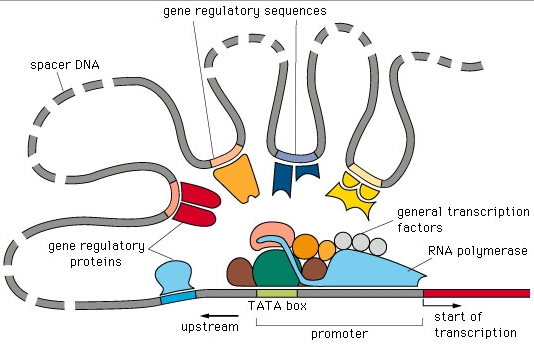
Indeed, for many years, work in the area concentrated
on finding the sequences upstream of genes that could correspond to promoter or other regulatory sequences, that is, to sites where protein, RNA or protein/RNA complexes would bind and
thereby initiate, stop, up or down-regulate the level of expression of a given gene. Even the cooperative
aspects of such binding 1, 2 was for long fully or partly ignored in the algorithms developed
to identify the sites from the genomic sequence alone 3, or, in rare cases, from the genomic sequence and its inferred structure
4. Such analyses were essentially based on the simplistic assumption that the sequence of regulatory sites should be more conserved on average than the remaining non-coding sequence.
The advent of high-throughput microarray technologies has added one important level of information to this image, as it enabled to measure the
co-expression of sets of genes in a given tissue in given conditions. These studies suggest a correlated action of different elements of the gene regulation machinery and their potential interaction, but
otherwise the image in the figure above remained basically the same, at least in the minds of
computational biologists.
More recently, the importance and extent of regulation at the epigenetic level was more fully
realised. This
refers to heritable changes in gene regulation that occur without a change in the DNA sequence and are therefore not encoded at the genomic level. Epigenetic regulation was nevertheless recognized first at the scale of DNA molecules. This
concerns in particular the chromatin structure and the possible chemical modification of some DNA bases as illustrated by the figure below.
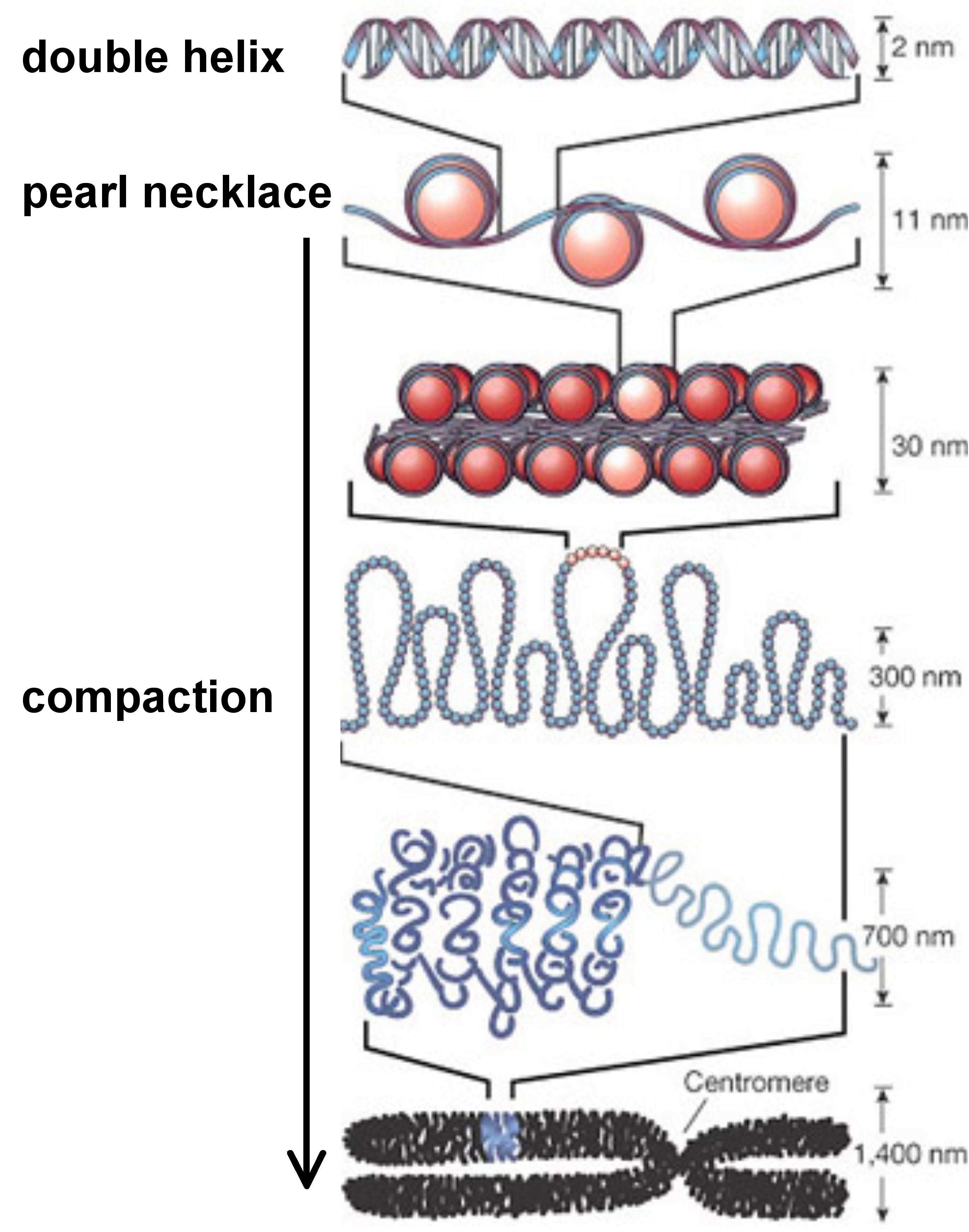
Chromatin is a complex made of DNA and of a special type of protein, called histones, around which DNA molecules densely wound to form a packed structure. Histones undergo post-translational modifications which alter their interaction with DNA and with proteins in the cell's nucleus. Combinations of such modifications are thought to constitute a code, the so-called ``histone code'' that plays a role in genome function, including gene regulation and DNA repair. The DNA itself can also be modified and this concerns mainly DNA methylation. In vertebrates, methylation typically occurs at CpG sites, that is, at sites where a cytosine (C) is directly followed by a guanine (G) in the DNA sequence. Such sites are often found at higher density near vertebrate gene promoter sequences where they are collectively referred to as CpG islands. Both chromatin structure and CpG islands have become an increasing concern
of computational biologists (whether coming from mathematics, physics and/or computer science)
although good models and data are often missing to be able to conduct large-scale exploratory computer studies.
While these aspects of epigenetic regulation remain still largely unexplored by computational biologists, another level of complexity has in the last two decades emerged in the study of gene regulation. It is indeed now increasingly realised that,
besides chromatin structure and DNA modification, the spatial arrangement of the chromosomes of a eukaryotic genome inside
a cell is related to gene regulation, and possibly also to other important life processes.
Such spatial arrangement, also called the nuclear architecture, is not random. During interphase, chromosomes occupy distinct territories with preferred radial locations inside the nucleus 5, that is with preferential locations relative to the nuclear center. This is illustrated in the figure below.
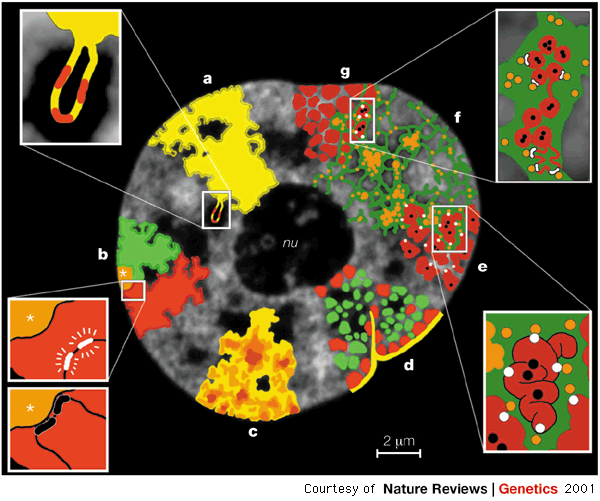
These preferred locations are cell type-specific and are conserved in the same cell type accross different primates.
Interphase is a step of the cell cycle during which chromosomes are transcriptionally active, and DNA replication occurs before the next cell division. Eukaryotic cells spend in general most of their time in interphase, during which the cell may or may not be growing and dividing. Intra-chromosomal interaction or simple spatial proximity between genetic elements situated at often distant positions along the genome are already known to be important for gene expression. Indeed, transcription for instance seems to be localised
within discrete regions that have been called ``transcription factories'' 6, 7. These are multifunctional supercomplexes able to process multiple, often distally located genes. Until very recently, it was assumed that such interactions could be intra-chromosomal only. Chromosome territories (denoted by CT) were thought to be separated from one another by an interchromatin space that, except in rare observed cases, rendered impossible any inter-chromosomal contact. Work dating from this year 8 however suggests, based upon strong evidence, that CTs intermingle as illustrated in the figure below
extracted from 8.
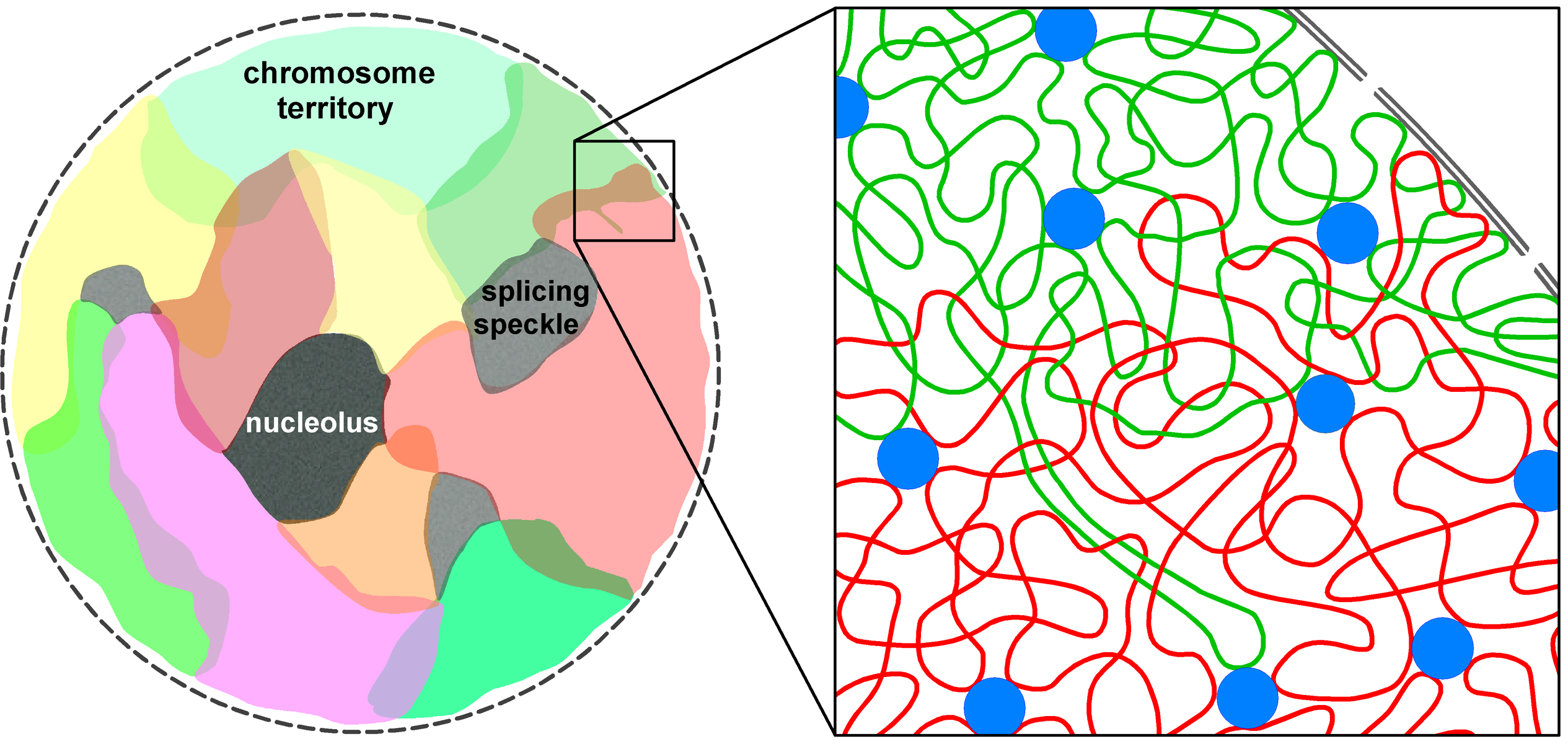
The pattern of intermingling has been shown to correlate with translocation frequencies (translocations are exchanges of genetic material between chromosomes). The pattern appears also to be changed when transcription is blocked. Furthermore, transcription factories have been identified in areas of intermingling
8. All those results, that will keep accumulating at an ever increasing speed as the community realises the value of the information held by the nuclear architecture, suggests
that inter-chromosomal interactions may take place at a much higher level than previously
thought. Indeed, some such interactions have been shown to exist in
a number of publications. Two that appeared this month in {\it Nature Genetics} 9, 10 describe new technologies that, together with other recent experimental techniques such as the one introduced in 8, enable already, for a given genomic locus, to determine the average frequency with which it interacts (by making contact) during interphase with other
loci, intra- or inter-chromosomes. Soon, it will be possible to obtain whole genome interaction matrices (A. Pombo, unpublished data). Inferring, from these observed frequencies of contact among genomic loci, even partially the network of all chromosomal interactions, in particular inter-chromosomal, may therefore be of essential importance. Indeed, it is expected that such network
is specific of the type of cell considered, as this would help explain
different whole chromosome conformations already identified as depending on cell type and gene activity.
Understanding this network may thus help better understand various life processes such as gene regulation, but also what makes two cells otherwise identical in terms of their genomic material different in their behaviour and role in differentiation and development.
Building the first computational and mathematical tools towards statistically and algorithmically exploring such network of intra- and inter-chromosomal interactions will be the main aim of this project.
References
[1] L. Marsan and M.-F. Sagot,
Algorithms for extracting structured motifs using a suffix tree with an application to promoter and regulatory site consensus identification, J Comput Biol., 7:345-362, 2000.
[2]
T. Werner, Models for prediction and recognition of eukaryotic promoters, Mamm Genome, 10:168-175, 1999.
[3] M. Tompa et al., Assessing computational tools for the discovery of transcription factor binding sites, Nat Biotechnol, 23:137-144, 2005.
[4] C. Deremble and R. Lavery, Macromolecular recognition, Curr Opin Struct Biol, 15:171-175, 2005.
[5] T. Cremer and C. Cremer, Chromosome territories, nuclear architecture and gene regulation in mammalian cells, Nat Rev Genet, 2:292-301, 2001.
[6] D.A. Jackson, A.B. Hassan, R.J. Herrington and P.R. Cook,
Visualization of focal sites of transcription within human nuclei,
EMBO J, 12:1059-1065, 1993.
[7] F.J Iborra, A. Pombo, D.A. Jackson and P.R. Cook, Active RNA polymerases are localized within discrete transcription "factories" in human nuclei, J Cell Sci, 109:1427-1436, 1996.
[8] M.R. Branco and A. Pombo, Intermingling of chromosome territories in interphase suggests role in translocations and transcription-dependent associations, PLoS Biol, 4:e138, 2006.
[9 M. Simonis, ..., and W. de Laat, Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C), Nat Genet, 38:1348-1354, 2006.
[10] Z. Zhao, ..., and R. Ohlsson, Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra- and interchromosomal interactions, Nat Genet, 38:1341-1347, 2006.