Static class providing methods to compute statistics on sequences data. More...
#include <Bpp/PopGen/SequenceStatistics.h>
Static Public Member Functions | |
| static unsigned int | numberOfPolymorphicSites (const PolymorphismSequenceContainer &psc, bool gapflag=true, bool ignoreUnknown=true) |
| Compute the number of polymorphic site in an alignment. More... | |
| static double | frequencyOfPolymorphicSites (const PolymorphismSequenceContainer &psc, bool gapflag=true, bool ignoreUnknown=true) |
| Compute the frequency of polymorphic site in an alignment. More... | |
| static unsigned int | numberOfParsimonyInformativeSites (const PolymorphismSequenceContainer &psc, bool gapflag=true) |
| Compute the number of parsimony informative sites in an alignment. More... | |
| static unsigned int | numberOfSingletons (const PolymorphismSequenceContainer &psc, bool gapflag=true) |
| Count the number of singleton nucleotides in an alignment. More... | |
| static unsigned int | totalNumberOfMutations (const PolymorphismSequenceContainer &psc, bool gapflag=true) |
| Count the total number of mutations in an alignment. More... | |
| static unsigned int | totalNumberOfMutationsOnExternalBranches (const PolymorphismSequenceContainer &ing, const PolymorphismSequenceContainer &outg) |
| Count the total number of mutations in external branches. More... | |
| static unsigned int | numberOfTriplets (const PolymorphismSequenceContainer &psc, bool gapflag=true) |
| Compute the number of triplet in an alignment. More... | |
| static double | heterozygosity (const PolymorphismSequenceContainer &psc, bool gapflag=true) |
| Compute the sum of per site heterozygosity in an alignment. More... | |
| static double | squaredHeterozygosity (const PolymorphismSequenceContainer &psc, bool gapflag=true) |
| Compute the sum of per site squared heterozygosity in an alignment. More... | |
| static double | gcContent (const PolymorphismSequenceContainer &psc) |
| Compute the mean GC content in an alignment. More... | |
| static std::vector< unsigned int > | gcPolymorphism (const PolymorphismSequenceContainer &psc, bool gapflag=true) |
| Return the number of GC alleles and the total number of alleles at polymorphic sites only. More... | |
| static double | watterson75 (const PolymorphismSequenceContainer &psc, bool gapflag=true, bool ignoreUnknown=true, bool scaled=false) |
| Compute diversity estimator Theta of Watterson (1975, Theor Popul Biol, 7 pp256-276) More... | |
| static double | tajima83 (const PolymorphismSequenceContainer &psc, bool gapflag=true, bool ignoreUnknown=true, bool scaled=false) |
| Compute diversity estimator Theta of Tajima (1983, Genetics, 105 pp437-460) More... | |
| static double | fayWu2000 (const PolymorphismSequenceContainer &psc, const Sequence &ancestralSites) |
| Compute diversity estimator Theta H (eq. 3) of Fay and Wu (2000, Genetics, 155: 1405-1413) More... | |
| static unsigned int | dvk (const PolymorphismSequenceContainer &psc, bool gapflag=true) |
| Return the number of haplotype in the sample. Depaulis and Veuille (1998, Mol Biol Evol, 12 pp1788-1790) More... | |
| static double | dvh (const PolymorphismSequenceContainer &psc, bool gapflag=true) |
| Return the haplotype diversity of a sample. Depaulis and Veuille (1998, Mol Biol Evol, 12 pp1788-1790) More... | |
| static unsigned int | numberOfTransitions (const PolymorphismSequenceContainer &psc) |
| Return the number of transitions. More... | |
| static unsigned int | numberOfTransversions (const PolymorphismSequenceContainer &psc) |
| Return the number of transversions. More... | |
| static double | ratioOfTransitionsTransversions (const PolymorphismSequenceContainer &psc) |
| Return the ratio of transitions/transversions. More... | |
| static unsigned int | numberOfSitesWithStopCodon (const PolymorphismSequenceContainer &psc, const GeneticCode &gCode, bool gapflag=true) |
| Compute the number of codon sites with stop codon. More... | |
| static unsigned int | numberOfMonoSitePolymorphicCodons (const PolymorphismSequenceContainer &psc, bool stopflag=true, bool gapflag=true) |
| Compute the number of polymorphic codon with only one mutated site. More... | |
| static unsigned int | numberOfSynonymousPolymorphicCodons (const PolymorphismSequenceContainer &psc, const GeneticCode &gc) |
| Compute the number of synonymous polymorphic codon sites. More... | |
| static double | watterson75Synonymous (const PolymorphismSequenceContainer &psc, const GeneticCode &gc) |
| Compute the Watterson(1975,Theor Popul Biol, 7 pp256-276) estimator for synonymous positions. More... | |
| static double | watterson75NonSynonymous (const PolymorphismSequenceContainer &psc, const GeneticCode &gc) |
| Compute the Watterson(1975, Theor Popul Biol, 7 pp256-276) estimator for non synonymous positions. More... | |
| static double | piSynonymous (const PolymorphismSequenceContainer &psc, const GeneticCode &gc, bool minchange=false) |
| Compute the synonymous nucleotide diversity, pi. More... | |
| static double | piNonSynonymous (const PolymorphismSequenceContainer &psc, const GeneticCode &gc, bool minchange=false) |
| Compute the non-synonymous nucleotide diversity, pi. More... | |
| static double | meanNumberOfSynonymousSites (const PolymorphismSequenceContainer &psc, const GeneticCode &gc, double ratio=1.) |
| compute the mean number of synonymous site in an alignment More... | |
| static double | meanNumberOfNonSynonymousSites (const PolymorphismSequenceContainer &psc, const GeneticCode &gc, double ratio=1.) |
| compute the mean number of non-synonymous site in an alignment More... | |
| static unsigned int | numberOfSynonymousSubstitutions (const PolymorphismSequenceContainer &psc, const GeneticCode &gc, double freqmin=0.) |
| compute the number of synonymous substitutions in an alignment More... | |
| static unsigned int | numberOfNonSynonymousSubstitutions (const PolymorphismSequenceContainer &psc, const GeneticCode &gc, double freqmin=0.) |
| compute the number of non synonymous substitutions in an alignment More... | |
| static std::vector< unsigned int > | fixedDifferences (const PolymorphismSequenceContainer &pscin, const PolymorphismSequenceContainer &pscout, PolymorphismSequenceContainer &psccons, const GeneticCode &gc) |
| compute the number of fixed differences between two alignements More... | |
| static std::vector< unsigned int > | mkTable (const PolymorphismSequenceContainer &ingroup, const PolymorphismSequenceContainer &outgroup, const GeneticCode &gc, double freqmin=0.) |
| return a vector containing Pa, Ps, Da, Ds More... | |
| static double | neutralityIndex (const PolymorphismSequenceContainer &ingroup, const PolymorphismSequenceContainer &outgroup, const GeneticCode &gc, double freqmin=0.) |
| return the neutrality index NI = (Pa/Ps)/(Da/Ds) (Rand & Kann 1996, Mol. Biol. Evol. 13 pp735-748) More... | |
| static double | tajimaDss (const PolymorphismSequenceContainer &psc, bool gapflag=true, bool ignoreUnknown=true) |
| Return the Tajima's D test (Tajima 1989, Genetics 123 pp 585-595). More... | |
| static double | tajimaDtnm (const PolymorphismSequenceContainer &psc, bool gapflag=true, bool ignoreUnknown=true) |
| Return the Tajima's D test (Tajima 1989, Genetics 123 pp 585-595). More... | |
| static double | fuLiD (const PolymorphismSequenceContainer &ingroup, const PolymorphismSequenceContainer &outgroup, bool useNbSingletons=true, bool useNbSegregatingSites=false) |
| Return the Fu and Li D test (Fu & Li 1993, Genetics, 133 pp693-709). More... | |
| static double | fuLiDStar (const PolymorphismSequenceContainer &group, bool useNbSegregatingSites=false) |
| Return the Fu and Li D* test (Fu & Li 1993, Genetics, 133 pp693-709). More... | |
| static double | fuLiF (const PolymorphismSequenceContainer &ingroup, const PolymorphismSequenceContainer &outgroup, bool useNbSingletons=true, bool useNbSegregatingSites=false) |
| Return the Fu and Li F test (Fu & Li 1993, Genetics, 133 pp693-709). More... | |
| static double | fuLiFStar (const PolymorphismSequenceContainer &group, bool useNbSegregatingSites) |
| Return the Fu and Li F* test (Fu & Li 1993, Genetics, 133 pp693-709). More... | |
| static double | fstHudson92 (const PolymorphismSequenceContainer &psc, size_t id1, size_t id2) |
| static std::unique_ptr< PolymorphismSequenceContainer > | generateLdContainer (const PolymorphismSequenceContainer &psc, bool keepsingleton=true, double freqmin=0.) |
| generate a special PolymorphismSequenceContainer for linkage disequilbrium analysis More... | |
| static Vdouble | pairwiseDistances1 (const PolymorphismSequenceContainer &psc, bool keepsingleton=true, double freqmin=0.) |
| give the vector of the pairwise distances between site positions corresponding to a LD SequencePolymorphismContainer More... | |
| static Vdouble | pairwiseDistances2 (const PolymorphismSequenceContainer &psc, bool keepsingleton=true, double freqmin=0.) |
| give the vector of all mean pairwise distance between two sites to a LD SequencePolymorphismContainer More... | |
| static Vdouble | pairwiseD (const PolymorphismSequenceContainer &psc, bool keepsingleton=true, double freqmin=0.) |
| give the vector of all mean pairwise D value between two sites (Lewontin & Kojima 1964, Evolution 14 pp458-472) More... | |
| static Vdouble | pairwiseDprime (const PolymorphismSequenceContainer &psc, bool keepsingleton=true, double freqmin=0.) |
| give the vector of all mean pairwise D' value between two sites (Lewontin 1964, Genetics 49 pp49-67)) More... | |
| static Vdouble | pairwiseR2 (const PolymorphismSequenceContainer &psc, bool keepsingleton=true, double freqmin=0.) |
| give the vector of all mean pairwise R² value between two sites (Hill & Robertson 1968, Theor. Appl. Genet., 38 pp226-231) More... | |
| static double | meanD (const PolymorphismSequenceContainer &psc, bool keepsingleton=true, double freqmin=0.) |
| give mean D over all pairwise comparisons More... | |
| static double | meanDprime (const PolymorphismSequenceContainer &psc, bool keepsingleton=true, double freqmin=0.) |
| give mean D' over all pairwise comparisons More... | |
| static double | meanR2 (const PolymorphismSequenceContainer &psc, bool keepsingleton=true, double freqmin=0.) |
| give mean R² over all pairwise comparisons More... | |
| static double | meanDistance1 (const PolymorphismSequenceContainer &psc, bool keepsingleton=true, double freqmin=0.) |
| give mean pairwise distances between sites / method 1: differences between sequences are not taken into account More... | |
| static double | meanDistance2 (const PolymorphismSequenceContainer &psc, bool keepsingleton=true, double freqmin=0.) |
| give mean pairwise distances between sites / method 2: differences between sequences are taken into account More... | |
| static double | originRegressionD (const PolymorphismSequenceContainer &psc, bool distance1=false, bool keepsingleton=true, double freqmin=0.) |
| give the slope of the regression |D| = 1+a*distance More... | |
| static double | originRegressionDprime (const PolymorphismSequenceContainer &psc, bool distance1=false, bool keepsingleton=true, double freqmin=0.) |
| give the slope of the regression |D'| = 1+a*distance More... | |
| static double | originRegressionR2 (const PolymorphismSequenceContainer &psc, bool distance1=false, bool keepsingleton=true, double freqmin=0.) |
| give the slope of the regression R² = 1+a*distance More... | |
| static Vdouble | linearRegressionD (const PolymorphismSequenceContainer &psc, bool distance1=false, bool keepsingleton=true, double freqmin=0.) |
| give the slope and the origin of the regression |D| = a*distance+b More... | |
| static Vdouble | linearRegressionDprime (const PolymorphismSequenceContainer &psc, bool distance1=false, bool keepsingleton=true, double freqmin=0.) |
| give the slope and the origin of the regression |D'| = a*distance+b More... | |
| static Vdouble | linearRegressionR2 (const PolymorphismSequenceContainer &psc, bool distance1=false, bool keepsingleton=true, double freqmin=0.) |
| give the slope and the origin of the regression R² = a*distance+b More... | |
| static double | inverseRegressionR2 (const PolymorphismSequenceContainer &psc, bool distance1=false, bool keepsingleton=true, double freqmin=0.) |
| give the slope of the regression R² = 1/(1+a*distance) More... | |
| static double | hudson87 (const PolymorphismSequenceContainer &psc, double precision=0.000001, double cinf=0.001, double csup=10000.) |
| give estimate of C=4Nr using Hudson method (Hudson 1987, Genet. Res., 50 pp245-250) More... | |
| static void | testUsefulValues (std::ostream &s, size_t n) |
| Test useful values. More... | |
Static Private Member Functions | |
| static unsigned int | getNumberOfMutations_ (const Site &site) |
| Count the number of mutation for a site. More... | |
| static unsigned int | getNumberOfSingletons_ (const Site &site) |
| Count the number of singleton for a site. More... | |
| static unsigned | getNumberOfDerivedSingletons_ (const Site &site_in, const Site &site_out) |
| Count the number of singleton for a site. More... | |
| static std::map< std::string, double > | getUsefulValues_ (size_t n) |
| Get useful values for theta estimators. More... | |
| static double | getVD_ (size_t n, double a1, double a2, double cn) |
| Get the vD value of equation (32) in Fu & Li 1993, Genetics, 133 pp693-709) More... | |
| static double | getUD_ (double a1, double vD) |
| Get the uD value of equation (32) in Fu & Li 1993, Genetics, 133 pp693-709) More... | |
| static double | getVDstar_ (size_t n, double a1, double a2, double dn) |
| Get the vD* value of D* equation in Fu & Li 1993, Genetics, 133 pp693-709) More... | |
| static double | getUDstar_ (size_t n, double a1, double vDs) |
| Get the uD* value of D* equation in Fu & Li 1993, Genetics, 133 pp693-709) More... | |
| static double | leftHandHudson_ (const PolymorphismSequenceContainer &psc) |
| give the left hand term of equation (4) in Hudson (Hudson 1987, Genet. Res., 50 pp245-250) This term is used in hudson87 More... | |
| static double | rightHandHudson_ (double c, size_t n) |
| give the right hand term of equation (4) in Hudson (Hudson 1987, Genet. Res., 50 pp245-250) This term is used in hudson87 More... | |
Detailed Description
Static class providing methods to compute statistics on sequences data.
Definition at line 34 of file SequenceStatistics.h.
Member Function Documentation
◆ dvh()
|
static |
Return the haplotype diversity of a sample. Depaulis and Veuille (1998, Mol Biol Evol, 12 pp1788-1790)
- Parameters
-
psc a PolymorphismSequenceContainer gapflag flag set by default to true if you don't want to take gaps into account
Definition at line 386 of file SequenceStatistics.cpp.
◆ dvk()
|
static |
Return the number of haplotype in the sample. Depaulis and Veuille (1998, Mol Biol Evol, 12 pp1788-1790)
- Parameters
-
psc a PolymorphismSequenceContainer gapflag flag set by default to true if you don't want to take gap into account
Definition at line 348 of file SequenceStatistics.cpp.
◆ fayWu2000()
|
static |
Compute diversity estimator Theta H (eq. 3) of Fay and Wu (2000, Genetics, 155: 1405-1413)
- Parameters
-
psc a PolymorphismSequenceContainer ancestralSites a Sequence containing the ancestral states (reconstructed independently) to fold the mutation in the psc SequenceContainer.
Definition at line 301 of file SequenceStatistics.cpp.
References bpp::TemplateVectorSiteContainer< class, class >::alphabet(), count(), bpp::Sequence::getChar(), bpp::TemplateVectorSiteContainer< class, class >::getNumberOfSites(), bpp::Alphabet::getSize(), bpp::Sequence::getValue(), bpp::TemplateVectorSiteContainer< class, class >::sequence(), bpp::TemplateVectorSiteContainer< class, class >::site(), and bpp::Sequence::size().
◆ fixedDifferences()
|
static |
compute the number of fixed differences between two alignements
Gaps and unresolved sites are automatically excluded
In case of complex codon, the path that gives the minimum number of non-synonymous changes is chosen. The argument minchange=true is sent to numberOfSynonymousDifferences used in this method. Otherwise, a non-integer number could be return.
- Parameters
-
pscin a PolymorphismSequenceContainer pscout a PolymorphismSequenceContainer psccons a PolymorphismSequenceContainer gc a GeneticCode
Definition at line 694 of file SequenceStatistics.cpp.
References bpp::CompleteTemplateSiteContainerIterator< class, class, class >::hasMoreSites(), and bpp::CompleteTemplateSiteContainerIterator< class, class, class >::nextSite().
◆ frequencyOfPolymorphicSites()
|
static |
Compute the frequency of polymorphic site in an alignment.
The number of polymorphic site is also known as the number of segregating site 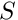 . This number is divided by the number of callable sites, which dependson the gapflag and ignoreUnknown arguments.
. This number is divided by the number of callable sites, which dependson the gapflag and ignoreUnknown arguments.
Gaps are consider as mutations so if you want number of polymorphic site without gap, set the gapflag parameter to true.
- Parameters
-
psc a PolymorphismSequenceContainer gapflag a boolean set by default to true if you don't want to take gap into account ignoreUnknown a boolean set by default to true to ignore unknown states
Definition at line 56 of file SequenceStatistics.cpp.
◆ fstHudson92()
|
static |
Fst of Hudson, Slatkin and Maddison
Taken from eq. 3 of Hudson, Slatkin and Maddison 1992 Genetics 132:153
![\[ F_{st} = 1 - \frac{H_w}{H_b} \]](form_22.png)
where 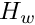 is mean number of differences between different sequences sampled from the same subpopulation, and
is mean number of differences between different sequences sampled from the same subpopulation, and 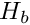 is the mean number of differences between sequences sampled from the two different subpopulations sampled.
is the mean number of differences between sequences sampled from the two different subpopulations sampled.
- Parameters
-
psc a PolymorphismSequenceContainer will at least two populations id1 is the id of the population 1 id2 is the id of the population 2
Definition at line 901 of file SequenceStatistics.cpp.
References bpp::TemplateVectorSiteContainer< class, class >::getNumberOfSites().
◆ fuLiD()
|
static |
Return the Fu and Li D test (Fu & Li 1993, Genetics, 133 pp693-709).
- Parameters
-
ingroup a PolymorphismSequenceContainer outgroup a PolymorphismSequenceContainer useNbSingletons use the original Fu & Li method based on the number of singletons, otherwise, use the total number of mutations in external branches. useNbSegregatingSites use the number of seggregating sites, otherwise use the total number of mutations. These two quantities are identical under the infinite site model, but the number of segregating sites will underestimate the total number of mutations in case of multiple substitutions at the same site.
- Exceptions
-
ZeroDivisionException if eta == 0
If one set original=false then the number of mutations will be used. If the outgroup contains more than one sequence the sites with more than one variant will not be considered for external branch mutations!
Definition at line 789 of file SequenceStatistics.cpp.
References bpp::TemplateVectorSiteContainer< class, class >::getNumberOfSequences().
◆ fuLiDStar()
|
static |
Return the Fu and Li D* test (Fu & Li 1993, Genetics, 133 pp693-709).
- Parameters
-
group a PolymorphismSequenceContainer useNbSegregatingSites use the number of seggregating sites, otherwise use the total number of mutations. These two quantities are identical under the infinite site model, but the number of segregating sites will underestimate the total number of mutations in case of multiple substitutions at the same site.
Definition at line 815 of file SequenceStatistics.cpp.
References bpp::TemplateVectorSiteContainer< class, class >::getNumberOfSequences().
◆ fuLiF()
|
static |
Return the Fu and Li F test (Fu & Li 1993, Genetics, 133 pp693-709).
- Parameters
-
ingroup a PolymorphismSequenceContainer outgroup a PolymorphismSequenceContainer external branch. useNbSingletons use the original Fu & Li method based on the number of singletons, otherwise, use the total number of mutations in external branches. useNbSegregatingSites use the number of seggregating sites, otherwise use the total number of mutations. These two quantities are identical under the infinite site model, but the number of segregating sites will underestimate the total number of mutations in case of multiple substitutions at the same site.
If one set original=false then the number of mutations will be used. If the outgroup contains more than one sequence the sites with more than one variant will not be considered for external branch mutations!
Definition at line 844 of file SequenceStatistics.cpp.
References bpp::TemplateVectorSiteContainer< class, class >::getNumberOfSequences().
◆ fuLiFStar()
|
static |
Return the Fu and Li F* test (Fu & Li 1993, Genetics, 133 pp693-709).
- Parameters
-
group a PolymorphismSequenceContainer useNbSegregatingSites use the number of seggregating sites, otherwise use the total number of mutations. These two quantities are identical under the infinite site model, but the number of segregating sites will underestimate the total number of mutations in case of multiple substitutions at the same site.
Definition at line 872 of file SequenceStatistics.cpp.
References bpp::TemplateVectorSiteContainer< class, class >::getNumberOfSequences().
◆ gcContent()
|
static |
Compute the mean GC content in an alignment.
- Parameters
-
psc a PolymorphismSequenceContainer
Definition at line 204 of file SequenceStatistics.cpp.
References bpp::TemplateVectorSiteContainer< class, class >::alphabet().
◆ gcPolymorphism()
|
static |
Return the number of GC alleles and the total number of alleles at polymorphic sites only.
G vs C and A vs T polymorphism are not taken into account
- Warning
- SG 15/03/2010: The code of this method is not clear. See implementation for more details.
- Parameters
-
psc a PolymorphismSequenceContainer gapflag a boolean set by default to true if you don't want to take gap into account
- Returns
- A std::vector of size 2 containing the number of GC alleles and the total number of alleles.
Definition at line 212 of file SequenceStatistics.cpp.
References bpp::TemplateVectorSiteContainer< class, class >::getNumberOfSequences().
◆ generateLdContainer()
|
static |
generate a special PolymorphismSequenceContainer for linkage disequilbrium analysis
Create a PolymorphismSequenceContainer with only polymorphic site : The value 1 is assigned to the most frequent allele, and 0 to the least frequent. This psc is needed to compute Linkage Disequilibrium Statistics. Should be used before excluding gaps, but sites with gaps are not counted as polymorphic sites. Singleton can be excluded. Polymorphic site with the lowest frequency < threshold can be excluded. Only polymorphic sites with 2 alleles are kept.
- Parameters
-
psc a PolymorphismSequenceContainer keepsingleton a boolean (true by default, false to exclude singleton) freqmin a float (to exclude site with the lowest allele frequency less than the threshold given by freqmin, 0 by default)
- Todo:
- To be moved to PolymorphismSequenceContainerTools.
Definition at line 943 of file SequenceStatistics.cpp.
References bpp::TemplateVectorSiteContainer< class, class >::getAlphabet(), bpp::TemplateVectorSiteContainer< class, class >::getNumberOfSites(), bpp::Site::getValue(), and bpp::TemplateVectorSiteContainer< class, class >::site().
◆ getNumberOfDerivedSingletons_()
|
staticprivate |
Count the number of singleton for a site.
will count singletons that are not in site_out (a site in outgroup) site_in is a site from an ingroup
Definition at line 1510 of file SequenceStatistics.cpp.
◆ getNumberOfMutations_()
|
staticprivate |
Count the number of mutation for a site.
Definition at line 1480 of file SequenceStatistics.cpp.
◆ getNumberOfSingletons_()
|
staticprivate |
Count the number of singleton for a site.
Definition at line 1497 of file SequenceStatistics.cpp.
◆ getUD_()
|
staticprivate |
Get the uD value of equation (32) in Fu & Li 1993, Genetics, 133 pp693-709)
- Parameters
-
a1 as describe in getUsefulValues vD as provided by getVD_
- Returns
- the uD value as double
Definition at line 1588 of file SequenceStatistics.cpp.
◆ getUDstar_()
|
staticprivate |
Get the uD* value of D* equation in Fu & Li 1993, Genetics, 133 pp693-709)
- Parameters
-
n the number of observed sequences a1 as describe in getUsefulValues vDs as provided by getVDstar_
- Returns
- the uD* value as double
Definition at line 1621 of file SequenceStatistics.cpp.
◆ getUsefulValues_()
|
staticprivate |
Get useful values for theta estimators.
- Parameters
-
n the number of observed sequences
- Returns
- A map with 11 values. Keys are a1, a2, a1n, b1, b2, c1, c2, cn, dn, e1 and e2. The values are :
![\[ a_1=\sum_{i=1}^{n-1}\frac{1}{i} \qquad a_2=\sum_{i=1}^{n-1}\frac{1}{i^2} \]](form_25.png)
![\[ a_{1n}=\sum_{i=1}^{n}\frac{1}{i} \]](form_26.png)
![\[ b_1=\frac{n+1}{3(n-1)} \qquad b_2=\frac{2(n^2+n+3)}{9n(n-1)} \]](form_27.png)
![\[ c_1=b_1-\frac{1}{a_1} \qquad c_2=b_2-\frac{n+2}{a_1n}+\frac{a_2}{a_1^2} \]](form_28.png)
![\[ c_n=2\frac{na_1-2(n-1)}{(n-1)(n-2)} \]](form_29.png)
![\[ d_n=c_n+\frac{n-2}{(n-1)^2}+\frac{2}{n-1}\left(\frac{3}{2}-\frac{2a_{1n}-3}{n-2}-\frac{1}{n}\right) \]](form_30.png)
where![\[ e_1=\frac{c_1}{a_1} \qquad e_2=\frac{c_2}{a_1^2+a_2} \]](form_31.png)
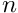 is the number of observed sequences.
is the number of observed sequences.
Definition at line 1532 of file SequenceStatistics.cpp.
◆ getVD_()
|
staticprivate |
Get the vD value of equation (32) in Fu & Li 1993, Genetics, 133 pp693-709)
- Parameters
-
n the number of observed sequences a1 as describe in getUsefulValues a2 as describe in getUsefulValues cn as describe in getUsefulValues
- Returns
- the vD value as double
Definition at line 1579 of file SequenceStatistics.cpp.
◆ getVDstar_()
|
staticprivate |
Get the vD* value of D* equation in Fu & Li 1993, Genetics, 133 pp693-709)
- Parameters
-
n the number of observed sequences a1 as describe in getUsefulValues a2 as describe in getUsefulValues dn as describe in getUsefulValues
- Returns
- the vD* value as double
Definition at line 1593 of file SequenceStatistics.cpp.
◆ heterozygosity()
|
static |
Compute the sum of per site heterozygosity in an alignment.
- Parameters
-
psc a PolymorphismSequenceContainer gapflag a boolean set by default to true if you don't want to take gap into account
Definition at line 167 of file SequenceStatistics.cpp.
◆ hudson87()
|
static |
give estimate of C=4Nr using Hudson method (Hudson 1987, Genet. Res., 50 pp245-250)
- Parameters
-
psc a PolymorphismSequenceContainer precision default value = 0.000001 cinf initial value, by default cinf=0.001 csup initial value, by default csup = 10000
Definition at line 1422 of file SequenceStatistics.cpp.
References bpp::TemplateVectorSiteContainer< class, class >::getNumberOfSequences().
◆ inverseRegressionR2()
|
static |
give the slope of the regression R² = 1/(1+a*distance)
To fit the theoretical prediction R² = 1/(1+4Nr) The slope is given in R² per kb
- Parameters
-
psc a PolymorphismSequenceContainer distance1 a boolean (true to use distance1, false to use distance2, false by default) keepsingleton a boolean (true by default, false to exclude singleton) freqmin a float (to exclude site with the lowest allele frequency less than the threshold given by freqmin, 0 by default)
- Exceptions
-
DimensionException if the number of sites or the number of sequences is lower than 2
Definition at line 1405 of file SequenceStatistics.cpp.
◆ leftHandHudson_()
|
staticprivate |
give the left hand term of equation (4) in Hudson (Hudson 1987, Genet. Res., 50 pp245-250) This term is used in hudson87
- Parameters
-
psc a PolymorphismSequenceContainer
Definition at line 1636 of file SequenceStatistics.cpp.
◆ linearRegressionD()
|
static |
give the slope and the origin of the regression |D| = a*distance+b
The slope is given in |D| per kb
- Parameters
-
psc a PolymorphismSequenceContainer distance1 a boolean (true to use distance1, false to use distance2, false by default) keepsingleton a boolean (true by default, false to exclude singleton) freqmin a float (to exclude site with the lowest allele frequency less than the threshold given by freqmin, 0 by default)
- Exceptions
-
DimensionException if the number of sites or the number of sequences is lower than 2
Definition at line 1363 of file SequenceStatistics.cpp.
◆ linearRegressionDprime()
|
static |
give the slope and the origin of the regression |D'| = a*distance+b
The slope is given in |D'| per kb
- Parameters
-
psc a PolymorphismSequenceContainer distance1 a boolean (true to use distance1, false to use distance2, false by default) keepsingleton a boolean (true by default, false to exclude singleton) freqmin a float (to exclude site with the lowest allele frequency less than the threshold given by freqmin, 0 by default)
- Exceptions
-
DimensionException if the number of sites or the number of sequences is lower than 2
Definition at line 1377 of file SequenceStatistics.cpp.
◆ linearRegressionR2()
|
static |
give the slope and the origin of the regression R² = a*distance+b
The slope is given in R² per kb
- Parameters
-
psc a PolymorphismSequenceContainer distance1 a boolean (true to use distance1, false to use distance2, false by default) keepsingleton a boolean (true by default, false to exclude singleton) freqmin a float (to exclude site with the lowest allele frequency less than the threshold given by freqmin, 0 by default)
- Exceptions
-
DimensionException if the number of sites or the number of sequences is lower than 2
Definition at line 1391 of file SequenceStatistics.cpp.
◆ meanD()
|
static |
give mean D over all pairwise comparisons
- Parameters
-
psc a PolymorphismSequenceContainer keepsingleton a boolean (true by default, false to exclude singleton) freqmin a float (to exclude site with the lowest allele frequency less than the threshold given by freqmin, 0 by default)
- Exceptions
-
DimensionException if the number of sites or the number of sequences is lower than 2
Definition at line 1296 of file SequenceStatistics.cpp.
◆ meanDistance1()
|
static |
give mean pairwise distances between sites / method 1: differences between sequences are not taken into account
- Parameters
-
psc a PolymorphismSequenceContainer keepsingleton a boolean (true by default, false to exclude singleton) freqmin a float (to exclude site with the lowest allele frequency less than the threshold given by freqmin, 0 by default)
- Exceptions
-
DimensionException if the number of sites is lower than 2
Definition at line 1314 of file SequenceStatistics.cpp.
◆ meanDistance2()
|
static |
give mean pairwise distances between sites / method 2: differences between sequences are taken into account
- Parameters
-
psc a PolymorphismSequenceContainer keepsingleton a boolean (true by default, false to exclude singleton) freqmin a float (to exclude site with the lowest allele frequency less than the threshold given by freqmin, 0 by default)
- Exceptions
-
DimensionException if the number of sites is lower than 2
Definition at line 1320 of file SequenceStatistics.cpp.
◆ meanDprime()
|
static |
give mean D' over all pairwise comparisons
- Parameters
-
psc a PolymorphismSequenceContainer keepsingleton a boolean (true by default, false to exclude singleton) freqmin a float (to exclude site with the lowest allele frequency less than the threshold given by freqmin, 0 by default)
- Exceptions
-
DimensionException if the number of sites or the number of sequences is lower than 2
Definition at line 1302 of file SequenceStatistics.cpp.
◆ meanNumberOfNonSynonymousSites()
|
static |
compute the mean number of non-synonymous site in an alignment
A site is x% synonymous if x% of possible mutations are synonymous The transition/transversion can be taken into account (use the variable ratio). Gaps are automatically excluded
- Parameters
-
psc a PolymorphismSequenceContainer gc a GeneticCode ratio a double
Definition at line 655 of file SequenceStatistics.cpp.
References bpp::CompleteTemplateSiteContainerIterator< class, class, class >::hasMoreSites(), and bpp::CompleteTemplateSiteContainerIterator< class, class, class >::nextSite().
◆ meanNumberOfSynonymousSites()
|
static |
compute the mean number of synonymous site in an alignment
A site is x% synonymous if x% of possible mutations are synonymous. The transition/transversion can be taken into account (use the variable ratio). Gaps and unresolved sites are automatically excluded.
- Parameters
-
psc a PolymorphismSequenceContainer gc a GeneticCode ratio a double
Definition at line 640 of file SequenceStatistics.cpp.
References bpp::CompleteTemplateSiteContainerIterator< class, class, class >::hasMoreSites(), and bpp::CompleteTemplateSiteContainerIterator< class, class, class >::nextSite().
◆ meanR2()
|
static |
give mean R² over all pairwise comparisons
- Parameters
-
psc a PolymorphismSequenceContainer keepsingleton a boolean (true by default, false to exclude singleton) freqmin a float (to exclude site with the lowest allele frequency less than the threshold given by freqmin, 0 by default)
- Exceptions
-
DimensionException if the number of sites or the number of sequences is lower than 2
Definition at line 1308 of file SequenceStatistics.cpp.
◆ mkTable()
|
static |
return a vector containing Pa, Ps, Da, Ds
Gaps and unresolved sites are automatically excluded
- Parameters
-
ingroup a PolymorphismSequenceContainer outgroup a PolymorphismSequenceContainer gc a GeneticCode freqmin a double, to exclude snp in frequency strictly lower than freqmin
Definition at line 720 of file SequenceStatistics.cpp.
References bpp::PolymorphismSequenceContainer::addSequence(), bpp::TemplateVectorSiteContainer< class, class >::getAlphabet(), bpp::TemplateVectorSiteContainer< class, class >::getNumberOfSequences(), bpp::TemplateVectorSiteContainer< class, class >::sequence(), and bpp::PolymorphismSequenceContainer::setAsOutgroupMember().
◆ neutralityIndex()
|
static |
return the neutrality index NI = (Pa/Ps)/(Da/Ds) (Rand & Kann 1996, Mol. Biol. Evol. 13 pp735-748)
Return -1 if Ps or Da are zero Gaps and unresolved sites are automatically excluded
- Parameters
-
ingroup a PolymorphismSequenceContainer outgroup a PolymorphismSequenceContainer gc a GeneticCode freqmin a double, to exclude snp in frequency strictly lower than freqmin
Definition at line 750 of file SequenceStatistics.cpp.
◆ numberOfMonoSitePolymorphicCodons()
|
static |
Compute the number of polymorphic codon with only one mutated site.
- Parameters
-
psc a PolymorphismSequenceContainer stopflag a boolean set by default to true if you don't want to take stop codon neither undefined sites into account gapflag a boolean set by default to true if you don't want to take gaps into account
Definition at line 553 of file SequenceStatistics.cpp.
◆ numberOfNonSynonymousSubstitutions()
|
static |
compute the number of non synonymous substitutions in an alignment
Gaps and unresolved sites are automatically excluded
In case of complex codon, the path that gives the minimum number of non-synonymous changes is chosen. The argument minchange=true is sent to numberOfSynonymousDifferences used in this method. Otherwise, a non-integer number could be return.
- Parameters
-
psc a PolymorphismSequenceContainer gc a GeneticCode freqmin a double, to exclude snp in frequency strictly lower than freqmin
Definition at line 682 of file SequenceStatistics.cpp.
References bpp::CompleteTemplateSiteContainerIterator< class, class, class >::hasMoreSites(), and bpp::CompleteTemplateSiteContainerIterator< class, class, class >::nextSite().
◆ numberOfParsimonyInformativeSites()
|
static |
Compute the number of parsimony informative sites in an alignment.
- Parameters
-
psc a PolymorphicSequenceContainer gapflag a boolean set by default to true if you don't want to take gap into account
Definition at line 77 of file SequenceStatistics.cpp.
◆ numberOfPolymorphicSites()
|
static |
Compute the number of polymorphic site in an alignment.
The number of polymorphic site is also known as the number of segregating site .
Gaps are consider as mutations so if you want number of polymorphic site without gap, set the gapflag parameter to true.
- Parameters
-
psc a PolymorphismSequenceContainer gapflag a boolean set by default to true if you don't want to take gap into account ignoreUnknown a boolean set by default to true to ignore unknown states
Definition at line 34 of file SequenceStatistics.cpp.
◆ numberOfSingletons()
|
static |
Count the number of singleton nucleotides in an alignment.
- Parameters
-
psc a PolymorphismSequenceContainer gapflag a boolean set by default to true if you don't want to take gap into account
Definition at line 96 of file SequenceStatistics.cpp.
◆ numberOfSitesWithStopCodon()
|
static |
Compute the number of codon sites with stop codon.
- Parameters
-
psc a PolymorphismSequenceContainer gCode the genetic code to use gapflag a boolean set by default to true if you don't want to take gaps into account
Definition at line 530 of file SequenceStatistics.cpp.
References bpp::TemplateVectorSiteContainer< class, class >::alphabet().
◆ numberOfSynonymousPolymorphicCodons()
|
static |
Compute the number of synonymous polymorphic codon sites.
Gaps and unresolved sites are automatically excluded
- Parameters
-
psc a PolymorphismSequenceContainer gc a GeneticCode
Definition at line 575 of file SequenceStatistics.cpp.
◆ numberOfSynonymousSubstitutions()
|
static |
compute the number of synonymous substitutions in an alignment
Gaps and unresolved sites are automatically excluded
In case of complex codon, the path that gives the minimum number of non-synonymous changes is chosen. The argument minchange=true is sent to numberOfSynonymousDifferences used in this method. Otherwise, a non-integer number could be return.
- Parameters
-
psc a PolymorphismSequenceContainer gc a GeneticCode freqmin a double, to exclude snp in frequency strictly lower than freqmin
Definition at line 669 of file SequenceStatistics.cpp.
References bpp::CompleteTemplateSiteContainerIterator< class, class, class >::hasMoreSites(), and bpp::CompleteTemplateSiteContainerIterator< class, class, class >::nextSite().
◆ numberOfTransitions()
|
static |
Return the number of transitions.
- Parameters
-
psc a PolymorphismSequenceContainer
Definition at line 433 of file SequenceStatistics.cpp.
◆ numberOfTransversions()
|
static |
Return the number of transversions.
- Parameters
-
psc a PolymorphismSequenceContainer
Definition at line 462 of file SequenceStatistics.cpp.
◆ numberOfTriplets()
|
static |
Compute the number of triplet in an alignment.
- Parameters
-
psc a PolymorphismSequenceContainer gapflag a boolean set by default to true if you don't want to take gap into account
Definition at line 112 of file SequenceStatistics.cpp.
◆ originRegressionD()
|
static |
give the slope of the regression |D| = 1+a*distance
The slope is given in |D| per kb
- Parameters
-
psc a PolymorphismSequenceContainer distance1 a boolean (true to use distance1, false to use distance2, false by default) keepsingleton a boolean (true by default, false to exclude singleton) freqmin a float (to exclude site with the lowest allele frequency less than the threshold given by freqmin, 0 by default)
- Exceptions
-
DimensionException if the number of sites or the number of sequences is lower than 2
Definition at line 1330 of file SequenceStatistics.cpp.
◆ originRegressionDprime()
|
static |
give the slope of the regression |D'| = 1+a*distance
The slope is given in |D'| per kb
- Parameters
-
psc a PolymorphismSequenceContainer distance1 a boolean (true to use distance1, false to use distance2, false by default) keepsingleton a boolean (true by default, false to exclude singleton) freqmin a float (to exclude site with the lowest allele frequency less than the threshold given by freqmin, 0 by default)
- Exceptions
-
DimensionException if the number of sites or the number of sequences is lower than 2
Definition at line 1341 of file SequenceStatistics.cpp.
◆ originRegressionR2()
|
static |
give the slope of the regression R² = 1+a*distance
The slope is given in R² per kb
- Parameters
-
psc a PolymorphismSequenceContainer distance1 a boolean (true to use distance1, false to use distance2, false by default) keepsingleton a boolean (true by default, false to exclude singleton) freqmin a float (to exclude site with the lowest allele frequency less than the threshold given by freqmin, 0 by default)
- Exceptions
-
DimensionException if the number of sites or the number of sequences is lower than 2
Definition at line 1352 of file SequenceStatistics.cpp.
◆ pairwiseD()
|
static |
give the vector of all mean pairwise D value between two sites (Lewontin & Kojima 1964, Evolution 14 pp458-472)
- Parameters
-
psc a PolymorphismSequenceContainer keepsingleton a boolean (true by default, false to exclude singleton) freqmin a float (to exclude site with the lowest allele frequency less than the threshold given by freqmin, 0 by default)
- Exceptions
-
DimensionException if the number of sites or the number of sequences is lower than 2
Definition at line 1160 of file SequenceStatistics.cpp.
References bpp::Site::getValue().
◆ pairwiseDistances1()
|
static |
give the vector of the pairwise distances between site positions corresponding to a LD SequencePolymorphismContainer
Assume that all sequences have the same length
- Parameters
-
psc a PolymorphismSequenceContainer keepsingleton a boolean (true by default, false to exclude singleton) freqmin a float (to exclude site with the lowest allele frequency less than the threshold given by freqmin, 0 by default)
- Exceptions
-
DimensionException if the number of sites is lower than 2
Definition at line 1015 of file SequenceStatistics.cpp.
References bpp::Site::alphabet(), bpp::TemplateVectorSiteContainer< class, class >::getNumberOfSites(), bpp::Alphabet::getSize(), and bpp::TemplateVectorSiteContainer< class, class >::site().
◆ pairwiseDistances2()
|
static |
give the vector of all mean pairwise distance between two sites to a LD SequencePolymorphismContainer
pairwise distances are computed for each sequence separately, excluding gaps. Then the mean is taken over all the sequences.
- Parameters
-
psc a PolymorphismSequenceContainer keepsingleton a boolean (true by default, false to exclude singleton) freqmin a float (to exclude site with the lowest allele frequency less than the threshold given by freqmin, 0 by default)
- Exceptions
-
DimensionException if the number of sites is lower than 2
Definition at line 1078 of file SequenceStatistics.cpp.
References bpp::Site::alphabet(), bpp::TemplateVectorSiteContainer< class, class >::getNumberOfSequences(), bpp::TemplateVectorSiteContainer< class, class >::getNumberOfSites(), bpp::Alphabet::getSize(), bpp::Sequence::getValue(), bpp::TemplateVectorSiteContainer< class, class >::sequence(), and bpp::TemplateVectorSiteContainer< class, class >::site().
◆ pairwiseDprime()
|
static |
give the vector of all mean pairwise D' value between two sites (Lewontin 1964, Genetics 49 pp49-67))
- Parameters
-
psc a PolymorphismSequenceContainer keepsingleton a boolean (true by default, false to exclude singleton) freqmin a float (to exclude site with the lowest allele frequency less than the threshold given by freqmin, 0 by default)
- Exceptions
-
DimensionException if the number of sites or the number of sequences is lower than 2
Definition at line 1196 of file SequenceStatistics.cpp.
References bpp::Site::getValue().
◆ pairwiseR2()
|
static |
give the vector of all mean pairwise R² value between two sites (Hill & Robertson 1968, Theor. Appl. Genet., 38 pp226-231)
- Parameters
-
psc a PolymorphismSequenceContainer keepsingleton a boolean (true by default, false to exclude singleton) freqmin a float (to exclude site with the lowest allele frequency less than the threshold given by freqmin, 0 by default)
- Exceptions
-
DimensionException if the number of sites or the number of sequences is lower than 2
Definition at line 1255 of file SequenceStatistics.cpp.
References bpp::Site::getValue().
◆ piNonSynonymous()
|
static |
Compute the non-synonymous nucleotide diversity, pi.
Gaps and unresolved sites are automatically excluded If minchange = false (default option) the different paths are equally weighted. If minchange = true the path with the minimum number of non-synonymous change is chosen.
- Parameters
-
psc a PolymorphismSequenceContainer gc a GeneticCode minchange a boolean set by default to false
Definition at line 625 of file SequenceStatistics.cpp.
References bpp::CompleteTemplateSiteContainerIterator< class, class, class >::hasMoreSites(), and bpp::CompleteTemplateSiteContainerIterator< class, class, class >::nextSite().
◆ piSynonymous()
|
static |
Compute the synonymous nucleotide diversity, pi.
Gaps and unresolved sites are automatically excluded If minchange = false (default option) the different paths are equally weighted. If minchange = true the path with the minimum number of non-synonymous change is chosen.
- Parameters
-
psc a PolymorphismSequenceContainer gc a GeneticCode minchange a boolean set to false
Definition at line 610 of file SequenceStatistics.cpp.
References bpp::CompleteTemplateSiteContainerIterator< class, class, class >::hasMoreSites(), and bpp::CompleteTemplateSiteContainerIterator< class, class, class >::nextSite().
◆ ratioOfTransitionsTransversions()
|
static |
Return the ratio of transitions/transversions.
- Parameters
-
psc a PolymorphismSequenceContainer
Definition at line 491 of file SequenceStatistics.cpp.
References count().
◆ rightHandHudson_()
|
staticprivate |
give the right hand term of equation (4) in Hudson (Hudson 1987, Genet. Res., 50 pp245-250) This term is used in hudson87
Definition at line 1660 of file SequenceStatistics.cpp.
◆ squaredHeterozygosity()
|
static |
Compute the sum of per site squared heterozygosity in an alignment.
- Parameters
-
psc a PolymorphismSequenceContainer gapflag a boolean set by default to true if you don't want to take gap into account
Definition at line 183 of file SequenceStatistics.cpp.
◆ tajima83()
|
static |
Compute diversity estimator Theta of Tajima (1983, Genetics, 105 pp437-460)
![\[ \hat{\theta}_\pi = \sum_{i=1}^{S} \left(1-\sum_{j=1}^{4} \frac{k_{j,i}\times\left(k_{j,i}-1\right)} {n_i\times\left(n_i-1\right)}\right) \qquad \textrm{with }k_{j,i}>0 \]](form_17.png)
where 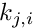 is the count of the jth state at the ith site,
is the count of the jth state at the ith site, 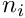 the number of nucleotides and the number of polymorphic sites.
the number of nucleotides and the number of polymorphic sites.
- Parameters
-
psc a PolymorphismSequenceContainer gapflag flag set by default to true if you don't want to take gap into account ignoreUnknown a boolean set by default to true to ignore unknown states scaled Tell if theta should be normalized per nucleotide (divided by the length of the sequence).
Definition at line 260 of file SequenceStatistics.cpp.
References count(), and bpp::TemplateVectorSiteContainer< class, class >::getAlphabet().
◆ tajimaDss()
|
static |
Return the Tajima's D test (Tajima 1989, Genetics 123 pp 585-595).
Calculation using the number of polymorphic (segregating) sites.
![\[ D=\frac{\hat{\theta}_\pi-\hat{\theta}_S}{\sqrt{\textrm{V}\left(\hat{\theta}_\pi-\hat{\theta}_S\right)}} =\frac{\hat{\theta}_\pi-\hat{\theta}_S}{\sqrt{e_1S+e_2S(S-1)}} \]](form_20.png)
- Parameters
-
psc a PolymorphismSequenceContainer gapflag flag set by default to true if you don't want to take gap into account ignoreUnknown a boolean set by default to true to ignore unknown states
- Exceptions
-
ZeroDivisionException if S == 0
Definition at line 763 of file SequenceStatistics.cpp.
References bpp::TemplateVectorSiteContainer< class, class >::getNumberOfSequences().
◆ tajimaDtnm()
|
static |
Return the Tajima's D test (Tajima 1989, Genetics 123 pp 585-595).
Calculation using the total number of mutation.
![\[ D=\frac{\hat{\theta}_\pi-\frac{\eta}{a_1}}{\sqrt{e_1\eta+e_2\eta(\eta-1)}} \]](form_21.png)
- Parameters
-
psc a PolymorphismSequenceContainer gapflag flag set by default to true if you don't want to take gap into account ignoreUnknown a boolean set by default to true to ignore unknown states
- Exceptions
-
ZeroDivisionException if eta == 0
Definition at line 776 of file SequenceStatistics.cpp.
References bpp::TemplateVectorSiteContainer< class, class >::getNumberOfSequences().
◆ testUsefulValues()
|
static |
Test useful values.
- Parameters
-
s a ostream where write the values n then number of observed sequences
Definition at line 1450 of file SequenceStatistics.cpp.
◆ totalNumberOfMutations()
|
static |
Count the total number of mutations in an alignment.
This count is assumed to be under an infinite site model.
- Parameters
-
psc a PolymorphismSequenceContainer gapflag a boolean set by default to true if you don't want to take gap into account
Definition at line 131 of file SequenceStatistics.cpp.
◆ totalNumberOfMutationsOnExternalBranches()
|
static |
Count the total number of mutations in external branches.
This is counted as the number of distinct singleton nucleotide in the ingroup that are not shared with the outgroup. A site is ignored if it contains more than one variant in the outgroup. A site is ignored if it contains unresolved variants or gaps.
- Parameters
-
ing a PolymorphismSequenceContainer the ingroup alignment outg a PolymorphismSequenceContainer the outgroup alignment
Definition at line 147 of file SequenceStatistics.cpp.
References bpp::TemplateVectorSiteContainer< class, class >::getNumberOfSites().
◆ watterson75()
|
static |
Compute diversity estimator Theta of Watterson (1975, Theor Popul Biol, 7 pp256-276)
![\[ \hat{\theta}_S=\frac{S}{a_1} \]](form_15.png)
where is the number of polymorphic sites and 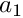 is describe in SequenceStatistics::getUsefulValues_().
is describe in SequenceStatistics::getUsefulValues_().
- Parameters
-
psc a PolymorphismSequenceContainer gapflag flag set by default to true if you don't want to take gap into account ignoreUnknown a boolean set by default to true to ignore unknown states scaled Tell if theta should be normalized per nucleotide (divided by the length of the sequence).
Definition at line 246 of file SequenceStatistics.cpp.
References bpp::TemplateVectorSiteContainer< class, class >::getNumberOfSequences().
◆ watterson75NonSynonymous()
|
static |
Compute the Watterson(1975, Theor Popul Biol, 7 pp256-276) estimator for non synonymous positions.
Gaps and unresolved sites are automatically excluded
In case of complex codon, the path that gives the minimum number of non-synonymous changes is chosen. The argument minchange=true is sent to numberOfSynonymousDifferences used in this method. Otherwise, a non-integer number could be return.
- Parameters
-
psc a PolymorphismSequenceContainer gc a GeneticCode
Definition at line 598 of file SequenceStatistics.cpp.
References bpp::TemplateVectorSiteContainer< class, class >::getNumberOfSequences().
◆ watterson75Synonymous()
|
static |
Compute the Watterson(1975,Theor Popul Biol, 7 pp256-276) estimator for synonymous positions.
Gaps and unresolved sites are automatically excluded
In case of complex codon, the path that gives the minimum number of non-synonymous changes* is chosen. The argument minchange=true is sent to numberOfSynonymousDifferences used in this method. Otherwise, a non-integer number could be return.
- Parameters
-
psc a PolymorphismSequenceContainer gc a GeneticCode
Definition at line 588 of file SequenceStatistics.cpp.
References bpp::TemplateVectorSiteContainer< class, class >::getNumberOfSequences().
The documentation for this class was generated from the following files:
- Bpp/PopGen/SequenceStatistics.h
- Bpp/PopGen/SequenceStatistics.cpp