Sommaire de la page
Structure résumée
Directeur du département: Laurent Duret DR CNRS
27 chercheurs et enseignants chercheurs permanents 2 personnes en CDD 4 post-doctorants et 24 doctorants
Génétique et Évolution des interactions Hôtes-Parasites
|
|
Anciens membres de l’équipe ayant quitté le laboratoire entre 2002 et 2005:
C. Lemaître MCU (retraite au 1/09/2005) ; D. Sillans MCU (retraite au 1/09/2004) ; J. Boulétreau-Merle CR1 CNRS (retraite au 1/01/2005) ; F. Dedeine (thèse 2003) MCU Tours ; L. Mouton (thèse 2004) Post-doc. Suisse ; L. Gavotte (thèse 2004), Post-doc. USA ; N. Ris (thèse 2003), IE INRA ; Bart Pannebakker (post doc CNRS 2004-2005).
Génomes et Populations
|
|
Anciens membres de l’équipe ayant quitté le laboratoire entre 2002 et 2005:
C. Rizzon (thèse 2003) MCU à Evry ; G. Decéliere (thèse 2004) post-doc au Cemagref de Montargis ; G. Zampicinini (thèse 2005) post-doc. à Torino.
BioInformatique et Génomique Évolutive
|
|
Anciens membres de l’équipe ayant quitté le laboratoire entre 2002 et 2005:
G. Bronner (thèse 2002) MCU à Clermont-Ferrand ; F. Thomarat (thèse 2002) MCU à Nancy ; L. Ponger (thèse 2003) MCU au MNHN ; J. Grassot (thèse 2004) ingénieur conseil ; A. Khelifi (thèse 2005) recherche d’emploi ; J-F. Dufayard (thèse 2005) IR contractuel CNRS, Montpellier ; J. Meunier (thèse 2005) recherche d’emploi ; M. Sémon (thèse 2005) Post-doc. à Dublin ; T. Silvestre (IR CNRS en CDD 2002-2004) formation complémentaire ; A. Urrutia (post-doc 2004-2005) post-doctorante en Angleterre.
Baobab
|
|
Anciens membres de l’équipe ayant quitté le laboratoire entre 2002 et 2005:
Nadia Pisanti (Post-doc 2001-2003) post-doctorante à l’Université de Pise ; Sébastien Provencher, (Post-doc 2001-2003) post-doctorant au Canada ; Arnaud Chaumot (thèse 2002) agrégé-préparateur ; Raquel Tavares (post-doc 2002-2003) MCU à Lyon1 ; Marina Zelwer (en thèse 2001-2003) IE à l’Université de Saint Etienne.
Présentation du département
La compréhension du fonctionnement et de l’évolution des organismes vivants implique d’étudier des niveaux d’organisation très différents : depuis la molécule jusqu’à l’écosystème, en passant par la cellule, l’organisme et la population. Ainsi, pour comprendre comment les espèces évoluent, comment elles s’adaptent à leur environnement, il est nécessaire de prendre en compte la dynamique des populations et les interactions entre les différentes espèces au sein d’un même écosystème. Mais il est aussi essentiel de comprendre quelles sont les bases moléculaires à l’origine de la variabilité phénotypique observée au sein des populations (quelles sont les mécanismes moléculaires responsables des mutations du matériel génétique ; quels sont les gènes qui sont impliqués dans la réponse adaptative). Par ailleurs, pour comprendre le fonctionnement du génome, il ne suffit pas de disséquer la fonction biochimique et l’expression des gènes qu’il contient : chaque génome est le fruit de milliards d’années d’évolution, et il est indispensable de prendre en compte l’histoire évolutive des génomes (et donc des espèces) pour pouvoir interpréter correctement les séquences génomiques contemporaines.
Les deux questions fondamentales de la biologie (comment fonctionne un organisme vivant ? et comment évolue-t-il ?) sont donc intrinsèquement liées. Pourtant, pendant longtemps ces différents aspects de la compréhension du monde vivant ont été étudiés au sein de disciplines relativement cloisonnées, centrées sur la biologie moléculaire d’une part, la génétique des populations d’autre part. Heureusement, depuis quelques années, et notamment depuis le démarrage des grands projets de séquençage de génomes, on assiste à un rapprochement entre ces disciplines. En effet, les techniques de production de données à haut débit (génome, transcriptome, protéome) constituent des outils extrêmement utiles aux biologistes des populations pour analyser la variabilité intraspécifique directement au niveau du support de l’information génétique. De plus, les progrès dans la connaissance des processus moléculaires (e.g. la recombinaison, la mutagénèse ou la réparation de l’ADN) ont permis une meilleure compréhension des facteurs qui influent sur cette variabilité intraspécifique. Par ailleurs, les biologistes moléculaires ont clairement réalisé que pour pouvoir décrypter ces séquences génomiques, il est indispensable d’étudier leur évolution. Ainsi, l’analyse de l’évolution des génomes (la génomique comparative) s’est avérée être une approche très efficace pour identifier les régions fonctionnelles (gènes, régions régulatrices) dans les séquences, et pour étudier les processus moléculaires du fonctionnement des génomes (réplication, transcription, traduction, recombinaison, réparation, mobilité des éléments transposables, ...).
Ce rapprochement entre biologie moléculaire et biologie des populations implique un investissement méthodologique fort pour gérer, manipuler, analyser et interpréter les grandes masses d’information : modélisation mathématique, développement d’outils statistiques et bioinformatiques.
Le département de « Génétique et Génomique Évolutives » a été constitué dans le but de développer une synergie entre des équipes de recherche recouvrant différentes disciplines complémentaires (génétique des populations, biologie moléculaire, génomique comparative, bioinformatique et biomathématiques), et visant à comprendre l’évolution et le fonctionnement des systèmes biologiques, en prenant en compte à la fois les paramètres populationnels et les mécanismes moléculaires.
Le département est découpé en quatre équipes, recouvrant différents champs disciplinaires :
- Génétique et Évolution des Interactions Hôtes-Parasites :
- Génétique des populations, biologie moléculaire, modélisation mathématique.
- Génomes et Populations :
- Génétique des populations, biologie moléculaire, bioinformatique, modélisation mathématique .
- BioInformatique et Génomique Évolutive :
- Évolution moléculaire, bioinformatique, statistiques, génétique des populations.
- Baobab :
- Évolution des génomes, des réseaux biologiques, algorithmique, modélisation, analyse de systèmes dynamiques.
Les principaux thèmes auxquels nous nous intéressons sont les suivants :
- Comment évoluent les organismes au sein de systèmes hôtes-parasites ? Quelles sont les bases génétiques des réponses adaptatives en considérant les différentes composantes des systèmes parasitaires et leurs interactions (l’hôte, le parasite, leur environnement biotique et abiotique) ?
- Quel rôle jouent les éléments transposables dans l’évolution des génomes et dans l’adaptation des espèces à leur environnement ? Quels sont les facteurs qui influent sur l’invasion des génomes par ces séquences répétées ?
- Quels sont les mécanismes moléculaires à l’origine de l’évolution des séquences génomiques et des répertoires de gènes ? Comment tirer parti de l’étude de l’évolution des séquences pour identifier les régions fonctionnelles dans les chromosomes ou pour mettre en évidence des processus moléculaires liés au fonctionnement du génome ?
- Comment reconstruire l’histoire évolutive des espèces à partir de l’analyse comparative des génomes (phylogénie moléculaire) ? Comment détecter les traces laissées par l’action de la sélection naturelle sur les séquences génomiques ?
- Comment évoluent les réseaux d’interaction moléculaires (e.g. voies métaboliques, réseaux de régulation, ...) ?
Parallèlement à ces grandes questions, nous consacrons une part très importante de notre recherche au développement d’outils mathématiques (notamment pour formaliser les questions posées), statistiques (pour analyser les données) et informatiques (développement de bases de données, d’algorithmes, etc.). Ces différents outils sont largement mis à la disposition de la communauté scientifique, notamment au travers de services web du Pôle Bioinformatique Lyonnais (PBIL).
Nous pensons qu’une des forces de notre département est qu’il regroupe des chercheurs provenant de formations très diverses (biologie moléculaire, génétique des populations, informatique, mathématiques). Cette variété de formations permet d’initier des travaux réellement pluridisciplinaires, en particulier via le co-encadrement de doctorants par des chercheurs de différentes équipes du département (7 étudiants co-encadrés au cours des quatre dernières années). Cette pluridisciplinarité se traduit également par le fait que deux des équipes du département (BioInformatique et Génomique Évolutive, Baobab) sont associées à l’INRIA (projet Hélix, regroupant également l’équipe d’Alain Viari, de l’INRIA Rhône-Alpes, Grenoble).
Enfin, il faut également souligner la bonne attractivité de notre département, qui a accueilli huit nouveaux chercheurs au cours de ces quatre dernières années : deux MCU (S. Mousset, J. Varaldi), quatre CR CNRS (V. Daubin, P. Gibert, E. Lerat, G. Marais), un CR INRIA (E. Tannier) et un DR INRA (D. Kahn, en détachement INRIA à partir de septembre 2005). Ces recrutements ont permis de développer de nouveaux sujets de recherche et de renforcer la pluridisciplinarité au sein de notre département en apportant de nouvelles compétences, notamment en algorithmique et en génétique des populations théorique.
Les travaux de recherche développés dans chacune de ces équipes sont présentés dans les chapitres suivants.
Génétique et Évolution des interactions Hôtes-Parasite
Les systèmes hôtes-parasites sont caractérisés par des interactions génétiques intenses qui vont de la modulation de l’expression des gènes de l’hôte par la présence d’un parasite jusqu’aux modifications de la composition génétique des populations sous l’action des interactions réciproques entre partenaires. Cette évolution peut conduire à des co-adaptations impliquant un grand nombre de caractères et responsables des principales caractéristiques des systèmes parasitaires (virulence, spécialisation, degré de dépendance, diversité des communautés). Nos recherches privilégient l’analyse génétique des interactions hôtes-parasites par une approche intégrative et fonctionnelle. Elles visent à identifier les supports génétiques des réponses adaptatives en considérant les différentes composantes des systèmes parasitaires et leurs interactions (l’hôte, le parasite, leur environnement biotique et abiotique, co-variation des caractères et trade-offs).
Cette approche a conduit à mettre en évidence l’importance des microorganismes symbiotiques (bactéries, virus) qui interviennent comme troisième partenaire dans les systèmes hôtes-parasites étudiés constitués des Drosophiles, leurs Hyménoptères parasitoïdes et leur cortège de micropartenaires procaryotes. Les bactéries Wolbachia font l’objet de plusieurs programmes de recherche compte tenu de leurs effets sur la reproduction de leur hôte. L’analyse de la variabilité génétique des populations, son organisation spatio-temporelle ainsi que sa signification adaptative sont complétées par l’étude de la relation génotype-phénotype et des interactions génotype-génotype-environnement (hôtes-parasite-facteurs abiotiques).
Les recherches se trouvent ainsi au carrefour des approches écologiques, génétiques et moléculaires de l’Evolution, et tentent une synthèse originale en développant à la fois études de terrain, expérimentations au laboratoire et théorie. Elles intègrent aussi des problèmes environnementaux liés à l’anthropisation des milieux (xénobiotiques, réchauffement climatique, espèces invasives), avec notamment des programmes sur les Trichogrammes, insectes parasitoïdes utilisés en contrôle biologique.
Biodiversité des communautés naturelles, phylogénie des hôtes, des parasitoïdes et de leurs micropartenaires symbiotiques
La connaissance approfondie des groupes taxonomiques composant les communautés étudiées est un souci permanent et un préalable à l’étude de l’évolution de ces systèmes, notamment afin d’identifier les forces sélectives qui agissent
(R. Allemand, M. Boulétreau, F. Vavre)
Trois nouvelles espèces parasitoïdes du genre Leptopilina ont été décrites par des méthodes complémentaires (morphologie, croisements, séquençage du gène ITS2 et caractérisation de leurs symbiotes), ce qui a permis de proposer la phylogénie du genre à l’échelle de l’Ancien Monde et une identification moléculaire des espèces par profils de restriction (Allemand et al., 2003). Le groupe L. heterotoma apparaît totalement infecté par des Wolbachia inductrices d’incompatibilité cytoplasmique, et la phylogénie comparée des hôtes et des parasites a révélé des transferts horizontaux de ces bactéries à hérédité maternelle. Le groupe L. clavipes est infecté par des Wolbachia induisant la parthénogenèse, et un événement de co-spéciation est suspecté. Enfin, le groupe L. boulardi est totalement indemne d’infection par Wolbachia, mais héberge un virus manipulateur du comportement (Varaldi et al., 2003). Ces résultats montrent que la dynamique évolutive des infections à Wolbachia est probablement influencée par les contraintes phylogénétiques des hôtes, et suggèrent des interactions entre microparasites.
2) Structuration spatiale et différenciation génétique des populations
L’organisation spatiale de la variabilité génétique des hôtes et des parasites est analysée à différentes échelles afin de déterminer quels caractères répondent à la sélection, quelle est leur implication dans le fonctionnement de la relation hôtes-parasites (adaptation locale face à quels facteurs sélectifs ?) et dans quelle mesure cette différenciation génétique contribue à la diversité des communautés.
(J. Boulétreau-Merle, P. Gibert, R. Allemand, F. Fleury, F. Vavre, J. Varaldi, M. Boulétreau, N. Ris, L. Mouton)
À l’échelle macrogéographique chez les drosophiles, l’espèce cosmopolite D. melanogaster a la capacité de se développer à la fois en région tempérée et tropicale suite à une différenciation génétique des populations que vient compléter une forte plasticité des caractères (Boulétreau-Merle et al., 2003). Une grande divergence génétique a été mesurée sur de très nombreux caractères physiologiques (ex : résistance au froid) et morphologiques (ex : nombre de soies sternopleurales, pigmentation, Gibert et al., 2004a). La température est considérée comme le principal facteur responsable de ces variations latitudinales. Par ailleurs, la plasticité phénotypique des caractères montre un net parallélisme avec les variations génétiques géographiques (Gibert et al., 2004b). Ceci est un argument en faveur d’une adaptation génétique à des conditions écologiques variables.
À l’échelle de la vallée du Rhône, le parasitoïde L. heterotoma montre une forte différenciation génétique Nord-Sud, les populations méditerranéennes ayant des valeurs phénotypiques plus élevées pour de nombreux caractères impliqués dans la fitness (fécondité, taille). Les analyses génétiques suggèrent que la sélection porte sur des gènes communs à effets pléiotropes synergiques (Ris, thèse 2003), ce qui devra être confirmé par une approche QTL. L’hypothèse explicative privilégiée est une réponse adaptative des populations méridionales aux pressions sélectives exercées par l’espèce compétitrice L. boulardi, qui supplante rapidement L. heterotoma dans la nature (Fleury et al., 2004). Un autre facteur d’adaptation et de différenciation est la température. Les réponses des génotypes nord et sud des parasitoïdes aux combinaisons température-espèce hôte sont en adéquation avec les conditions climatiques et écologiques auxquelles ils sont habituellement soumis. Ces interactions génotype-environnement confirment la valeur adaptative de la différenciation (Ris et al. 2004), mais ne sont pas suffisantes pour expliquer son maintien compte tenu des flux géniques importants mesurés par marqueurs microsatellites. Les populations pourraient ne pas être à l’équilibre sélection-migration suite à une réponse adaptative récente des populations méditerranéennes de L. heterotoma, peut être liée à une augmentation des températures à l’origine de l’implantation et la progression vers le nord de L. boulardi (espèce invasive ?). La spécificité et le niveau de co-adaptation des partenaires sont maintenant abordés par une approche protéomique afin d’identifier les déterminants moléculaires et génétiques impliqués (programme de P. Gibert).
Les interactions insectes-Wolbachia montrent également des variations géographiques en ce qui concerne la densité en bactéries, soumise à un compromis évolutif entre maintenir une densité suffisante assurant une transmission verticale efficace, tout en limitant le coût de l’infection sur l’insecte hôte (Mouton et al., 2004). La température de développement des insectes module sensiblement la densité bactérienne intracellulaire, et cet effet varie suivant le génotype et l’origine géographique de l’hôte (Mouton et al., 2005). Ces résultats sont à mettre en relation avec la prévalence de l’infection dans la nature, où les populations les plus septentrionales semblent présenter une diminution de l’infection durant la période hivernale. La forte densité bactérienne de ces génotypes à faible température leur permettrait de limiter les risques de perte de l’infection durant l’hiver. Ces hypothèses devront être confirmées par l’analyse quantitative de la transmission bactérienne sous différentes combinaisons génotype × température.
3) Réponse des populations aux facteurs d’origine anthropique
Les recherches sur les réponses adaptatives des populations intègrent également les facteurs d’origine anthropique comme source de perturbations phénotypiques et comme contraintes évolutives. Il s’agit de comprendre comment les organismes ectothermes comme les insectes peuvent répondre à des changements environnementaux liés aux activités humaines (augmentation des températures ou présence de xénobiotiques).
(JM Delpuech, R. Allemand, C. Dupond)
La limite nord du parasitoïde L. boulardi est imposée par les exigences thermiques de l’espèce et il est possible de tester l’hypothèse d’un réchauffement en suivant sa progression (une variation d’un degré correspond à une distance de 100 km). Depuis deux ans, l’aire de répartition de L. boulardi a effectivement progressé d’une cinquantaine de kilomètres, ce qui risque à terme de modifier l’équilibre des communautés dans cette zone. L’étude de ces communautés sera poursuivie sur le long terme, car elle offre des conditions exceptionnelles pour l’évaluation très fine des conséquences génétiques (sur L. heterotoma) et écologiques (sur l’ensemble de la communauté) des changements climatiques en cours.
Les insecticides constituent une pollution environnementale à laquelle les populations naturelles doivent s’adapter. Les hyménoptères parasitoïdes figurent parmi les principaux régulateurs naturels des populations d’insectes et sont donc des espèces clefs des écosystèmes. Nous avons montré l’impact des faibles doses d’insecticides sur les différentes phases de la reproduction des parasitoïdes des genres Trichogramma et Leptopilina. Les insecticides provoquent par exemple une diminution de la reconnaissance spécifique des femelles par les mâles, qui peut augmenter les tentatives d’accouplements interspécifiques et entraîner une forte diminution de la valeur sélective des femelles (Dupont, thèse en cours). À l’inverse, les insecticides peuvent augmenter l’attraction exercée par les kairomones de l’hôte sur les parasitoïdes (Delpuech et al., 2005), ce qui peut représenter un avantage dans une situation naturelle où l’hôte serait rare. Ces résultats démontrent la nécessité d’une étude très complète de l’effet des xénobiotiques pour déterminer leur impact réel au niveau des écosystèmes.
4) Déterminants symbiotiques du phénotype et de sa variabilité
Les insectes présentent la particularité d’héberger de très nombreux micro-organismes symbiotiques, qui participent activement au phénotype de leur hôte (phénotype étendu) et agissent comme pression de sélection pouvant interférer avec les autres contraintes de l’environnement. L’effet de ces partenaires symbiotiques et leurs conséquences démographiques et génétiques sont analysés.
(F. Vavre, J. Varaldi, F. Fleury, M. Boulétreau, F. Dedeine, L. Mouton, L. Gavotte, E. Vautrin, N. Kremer)
Un des résultats les plus originaux a été la découverte qu’un virus jusqu’alors inconnu modifie le comportement des femelles parasitoïdes. Chez l’espèce L. boulardi, la décision d’accepter ou de rejeter un hôte déjà parasité montre deux phénotypes extrêmes, l’un superparasitant (dépôt de plusieurs œufs dans le même hôte), l’autre non superparasitant. La transmission de ce comportement est à la fois maternelle et infectieuse au sein d’une larve de drosophile superparasitée. La mise en évidence de cette transmission horizontale, combinée à un travail de microscopie électronique, a permis de démontrer la responsabilité d’un virus filamenteux dans le phénotype superparasitant (Varaldi et al., 2003) (Figure 1). Ce virus présente une très faible pathogénicité physiologique sur la plupart des autres caractères (survie, taille, durée de développement) et cause un surprenant effet positif sur le stock d’œufs (Varaldi et al., 2005). La découverte de ce virus a remis en question l’interprétation adaptative du superparasitisme chez les parasitoïdes (Gandon et al., 2005).
Les Wolbachia, bactéries manipulatrices de la reproduction, montrent une forte variabilité de leurs effets entre espèces d’insectes, notamment pour l’incompatibilité cytoplasmique (Vavre et al., 2002 ; Dedeine et al., 2004), avec des répercussions directes sur la dynamique d’invasion et l’évolution de l’association (Vavre et al., 2003). La diversité et la densité bactérienne intracellulaire semblent constituer un déterminant important de la virulence de la bactérie (Mouton et al., 2004 et 2005) et la recherche des déterminants des effets de Wolbachia a conduit à écarter l’implication possible de leurs phages, dont la découverte récente pouvait laisser supposer l’implication dans la diversité mal expliquée des effets de la bactérie. L’étude de l’infection phagique de 38 souches de Wolbachia a montré une forte prévalence des phages dans les peuplements bactériens, mais une absence de relation entre phylogénie des phages et effets des bactéries (Gavotte et al., 2004). Le thème prometteur actuellement poursuivi dans l’équipe concerne l’implication de Wolbachia dans la capacité reproductive des femelles du parasitoïde A. tabida. Cette espèce est totalement dépendante d’une souche particulière de Wolbachia pour sa fécondité (Dedeine et al., 2004), créant une association obligatoire entre insecte et bactérie. Une transition entre association facultative et obligatoire semble exister puisqu’une forte variabilité a été observée entre des femelles totalement dépendantes de Wolbachia pour leur ovogenèse et d’autres ayant conservé une certaine autonomie (50% du stock d’ovocyte normal ; Dedeine et al., 2005). L’établissement des bases génétiques de cette variabilité se poursuit par l’étude cytologique précise du phénotype ovarien de l’hôte avec comme résultat prometteur le fait qu’en absence de Wolbachia les cellules nourricières entreraient en apoptose précoce (B. Pannebakker, post doc CNRS). Cette étude est complétée par la recherche des loci impliqués dans la variabilité, notamment ceux potentiellement impliqués dans les phénomènes d’apoptose, en étudiant leur niveau d’expression chez des individus infectés ou non infectés (thèse N. Kremer). Par ailleurs, grâce aux collaborations mises en place avec l’UMR 5557, l’analyse du génome de la souche de Wolbachia impliquée dans l’ovogenèse est en cours.
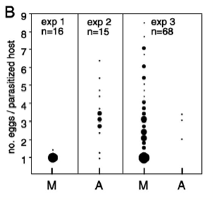
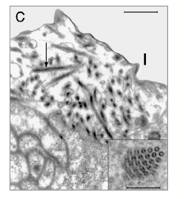
Figure 1 : Déterminisme viral du comportement de superparasitisme chez L. boulardi. Partie B : La figure illustre le nombre moyen d’oeufs que déposent les femelles de différentes lignées de L. boulardi dans leur hôte (larve de drosophile), la taille du cercle étant proportionnelle au nombre de femelles ayant le même phenotype. Les femelles de la population de Madère (M) déposent systématiquement un oeuf par hôte (exp1) alors que les femelles de la population d’Antibes (A) superparasitent fréquemment puisqu’un même hôte peut contenir jusqu’à 7 parasitoïdes alors qu’un seul survivra (exp 2). Lorsque les deux lignées se sont développées en compétition au sein du même hôte (exp 3), les femelles de Madère se mettent à superparasiter autant que celles d’Antibes, démontrant ainsi le caractère infectieux du phénotype « superparasitant ». Partie C : Photo en microscopie électronique du virus responsable de cette modification du comportement de superparasitisme (barre=500nm, encart 200nm). (Varaldi et al. 2003, Science).
5) Dynamique évolutive et épidémiologie des microparasites endosymbiotiques
Les microorganismes symbiotiques présentent souvent une transmission verticale incomplète, et leur maintien impose à chaque génération une contrainte à l’origine de leurs effets sur l’hôte. La diffusion de ces micro-partenaires est analysée en fonction de leur mode de transmission et de leurs effets, et des études théoriques essaient de comprendre comment peut évoluer le couple hôte- symbiote.
(F. Vavre, J. Varaldi, L. Mouton, E. Vautrin, S. Patot)
L’évolution et le maintien des multi-infections sont étudiés sur le modèle Wolbachia où les multi-infections sont fréquentes, malgré le goulot d’étranglement récurrent survenant lors de la colonisation des ovocytes. Des mécanismes actifs de maintien des multi-infections, comme l’incompatibilité cytoplasmique, induisent des pressions de sélection favorisant la transmission conjointe des bactéries et une éventuelle coopération entre souches. L’analyse des relations entre densité bactérienne et diversité intra-hôte a montré que la densité intracellulaire est régulée à un niveau spécifique, quelle que soit la diversité du peuplement bactérien (Mouton et al., 2004). L’absence de compétition entre souches pourrait limiter les pertes stochastiques de certaines bactéries et favoriser leur transmission conjointe. Les études en cours analysent les pressions sélectives qui agissent dans ces systèmes complexes, et cherchent à vérifier si la coopération entre souches peut émerger (thèse E. Vautrin).
L’épidémiologie et l’impact évolutif du virus inducteur du comportement de superparasitisme sont également analysés grâce à des modèles permettant de définir la stratégie optimale de superparasitisme de chacun des partenaires (collaboration avec Sylvain Gandon UMR CNRS – IRD Montpellier). Dans la grande majorité des situations écologiques testées, un conflit d’intérêt émerge (le virus est sélectionné pour induire plus de superparasitisme que le parasitoïde), ce qui suggère le caractère adaptatif de la modification du comportement du point de vue du virus. Lorsque les deux partenaires peuvent évoluer conjointement (coévolution), le conflit d’intérêt devient encore plus important. Le test de ces prédictions est en cours (thèse S. Patot).
6) Perspectives
Les programmes de recherche, tous centrés sur les relations hôtes-parasites, privilégieront une approche intégrative en maintenant une étroite connexion entre l’analyse de la variabilité génétique des populations et le contexte environnemental dans lequel elles vivent (quels caractères impliqués dans quelles adaptations et vis-à-vis de quels facteurs sélectifs). La maturité de certains programmes nécessite maintenant de développer une approche plus fonctionnelle à l’échelle de l’organisme afin de comprendre les mécanismes des interactions entre partenaires (gènes impliqués, modification de leur expression etc). Enfin, la recherche d’un modèle biologique complémentaire pouvant s’intégrer à l’ensemble de nos thématiques, mais apportant sa propre originalité et avec potentiellement de nouveaux axes scientifiques, a trouvé son aboutissement par la décision de travailler sur une espèce du genre Bemisia et son cortège parasitaire (virus, bactéries, parasitoïdes). L’importance agronomique de cette espèce est majeure en France et des collaborations avec des chercheurs de l’INRA et de pays étrangers sont en train de se mettre en place.
Bases génétiques des adaptations et relation génotype-phénotype
Les variations génétiques observées entre populations exposées à des conditions environnementales contrastées, et la co-variation de certains caractères, permettent d’envisager l’existence de gènes communs contrôlant plusieurs traits au départ supposés indépendants (effets pléiotropes). Les travaux chercheront à identifier ces gènes (approche QTL) afin de mieux comprendre la réponse génétique des populations face aux contraintes génétiques et environnementales auxquelles elles sont soumises (échelle spatio-temporelle, maintien de la diversité génétique). Un autre aspect privilégié sera l’étude de la spécialisation des populations vis à vis de leur environnement local, ce qui nécessite de mieux comprendre le lien entre génotype et phénotype via les interactions génotype-génotype-environnement qui sont une particularité des systèmes hôtes-parasites. Il s’agit de mesurer le degré de co-adaptation des partenaires face aux variations des facteurs abiotiques, que ce soit chez les insectes hôtes et leurs parasitoïdes ou entre insectes et leurs micro-organismes symbiotiques (virus, bactéries). Une approche protéomique est envisagée afin d’étudier les modifications sur l’hôte de la présence d’un parasite (gènes impliqués et leur niveau d’expression).
Enfin, la situation de la vallée du Rhône est un modèle idéal pour étudier le rôle des changements climatiques sur la dynamique de la biodiversité et la réponse adaptative des populations. Elle permet de comparer des espèces pouvant montrer des réponses différentes : soit une aire de répartition limitée par la température (L. boulardi absente au nord de Lyon) soit une aire de répartition large, mais dont les populations semblent localement adaptées aux conditions thermiques (L. heterotoma). Un suivi temporel des populations, initié depuis plusieurs années, permettra de déterminer comment ces espèces et leurs populations font face à des changements de température (réponse par plasticité phénotypique ou adaptation génétique). L’étude des effets des facteurs anthropiques concernera également les insecticides dont les doses sublétales peuvent profondément modifier le fonctionnement des systèmes étudiés.
Virus symbiotiques manipulateurs du comportement des insectes parasites : caractérisation, effets phénotypiques, conséquences démographiques et génétiques
La découverte qu’un virus, à transmission verticale et horizontale, peut induire le superparasitisme en modifiant le comportement de son hôte bouleverse notre vision du comportement des insectes, mais aussi celle des virus qui peuvent infecter de façon silencieuse les populations, voire devenir mutualistes sous certaines conditions. L’originalité de ce résultat nécessite de concentrer des efforts importants pour mieux comprendre cette association insecte-virus, tant au niveau de la diversité des relations que de la conséquence de ces infections virales sur la dynamique démographique et évolutive des systèmes hôte-parasitoïde. Les recherches auront pour objet l’étude des mécanismes d’action de ce virus (spécificité des effets, coût de l’infection, niveau d’altération du comportement, taux de transmission horizontaux et verticaux), de généraliser ce phénomène à d’autres espèces parasitoïdes, et de mettre au point les outils moléculaires nécessaires pour certains aspects fonctionnels, épidémiologiques et évolutifs des interactions virus-parasitoïdes. La distribution et les effets de ces virus seront recherchés dans d’autres systèmes hôte-parasitoide, qui hébergent des particules virales semblables et dont les effets sont encore inconnus. Une attention particulière sera portée aux conséquences de l’infection sur la composition génétique des populations infectées, notamment l’émergence possible de phénomènes de résistance dans les populations à forte prévalence. Il est également envisagé d’évaluer si ces infections virales sont importantes à prendre en compte dans l’efficacité des parasitoïdes comme agents de contrôle biologique des populations d’insectes phytophages. Ce programme de recherche fait l’objet d’un soutien par une ACI jeune chercheur qui fédère plusieurs membres du laboratoire.
Déterminants génétiques de la dépendance à Wolbachia pour l’ovogenèse chez A. tabida
L’espèce A. tabida où Wolbachia est devenue nécessaire à l’ovogenèse de l’insecte constitue une situation rare et idéale pour étudier la transition parasitisme-mutualisme et comprendre les processus à l’origine de la dépendance entre espèces et plus généralement l’évolution des relations hôtes-parasites. Les processus évolutifs impliqués seront analysés, et les mécanismes moléculaires de l’association abordés par une approche plus fonctionnelle de cette interaction. L’ovogenèse semble perturbée par l’entrée en apoptose précoce des cellules nourricières chez les individus non infectés ce qui nous conduit à entreprendre deux types d’approches (collaboration avec UMR 5557 Écologie Microbienne).
Une approche généraliste sera envisagée avec plusieurs étapes : (i) Cartographie QTL des gènes impliqués dans la dépendance chez A. tabida afin de déterminer le nombre de loci impliqués et leur localisation (ii) Obtention de séquences de Wolbachia voire séquençage total du variant et génomique comparative des Wolbachia (iii) recherche des protéines excrétées par Wolbachia.
La seconde approche sera ciblée sur l’apoptose. Des analyses cytologiques essayeront de confirmer l’existence d’apoptose au niveau ovarien chez les individus non infectés et de rechercher la spécificité tissulaire du phénomène. La machinerie apoptotique sera étudiée chez A. tabida en recherchant des homologues de gènes connus chez d’autres espèces, puis leur fonctionnalité et leur niveau d’expression avec ou sans Wolbachia seront testées. Enfin des gènes candidats chez Wolbachia seront recherchés et leur activité pro ou anti-apoptotique testée.
Réponse adaptative et diversité symbiotique chez Bemisia tabaci
Ce programme de recherche n’est actuellement qu’à l’état de projet, mais il se concrétise par le recrutement d’un premier étudiant sur le sujet (G. Gueguen M2) et le dépôt de 2 projets de recherche (1 national et 1 international). B tabaci constitue un modèle complémentaire de ceux étudiés actuellement, à fort potentiel à la fois scientifique et agronomique. Sa particularité est d’héberger un symbiote primaire obligatoire (complément nutritif pour des insectes piqueurs suceurs de sève phloemienne), qui coexiste avec de très nombreux symbiotes bactériens secondaires capables de modifier la reproduction de leurs hôtes (6 recensés dont Wolbachia) et dont les effets sont inconnus chez cette espèce. Par ailleurs, B. tabaci est vecteur de virus phytopathogènes dont la transmission et l’épidémiologie peuvent être fortement influencées par le compartiment bactérien. Le projet a pour objectifs d’analyser les interactions entre les partenaires symbiotiques via les effets qu’ils induisent sur leur hôte (interactions symbiotes primaire-secondaires, interactions bactéries-virus) et d’analyser la conséquence de ces infections sur la biologie et la dynamique invasive de B. tabaci. Il est ainsi dans le prolongement direct des travaux sur les parasitoïdes de drosophiles mais avec des particularités permettant d’envisager de nouveaux axes de recherche.
Génomes et Populations
1) Dynamique des éléments transposables chez les métazoaires
Alors que les gènes sont depuis longtemps l’objet d’intenses recherches, les séquences répétées, qui représentent souvent la majorité de l’ADN ne font l’objet d’un intérêt soutenu que depuis la prise de conscience que le génome humain porte 55% de telles séquences, et que ces séquences seraient en fait la clé des systèmes de régulation génique ainsi que de puissants facteurs d’évolution des génomes. Parmi ces séquences répétées, les éléments transposables présentent un intérêt particulier grâce à leur capacité de transposition, qui leur confère la possibilité d’envahir les génomes. Ils interviennent ainsi dans la taille des génomes, rendant compte du paradoxe de la valeur C selon lequel la taille des génomes n’est pas liée à la complexité des organismes. Ils jouent un rôle important comme agents mutateurs, interviennent dans la régulation de l’expression des gènes, et sont ainsi responsables d’un certain nombre de maladies. On ne peut comprendre l’évolution de la composition, de la structure et du fonctionnement des génomes qu’à la lumière d’une compréhension des facteurs responsables de la dynamique des séquences répétées et particulièrement des éléments transposables. Notre équipe aborde ces questions par le biais de la génétique des populations (dynamique du nombre de copies d’ET dans les populations naturelles) associée à la biologie moléculaire (structure et séquences des copies), la bioinformatique (analyse des copies d’ET dans les génomes séquencés) et la modélisation mathématique (modélisation des processus d’invasion d’ET dans les populations). Le modèle majeur reste la drosophile, mais l’anophèle constitue de plus en plus un modèle à priviligier, d’une part comme élément de comparaison avec la drosophile mais aussi parce que c’est un vecteur du paludisme.
Tandis que l’espèce Drosophila melanogaster a été longtemps le modèle priviligié des études sur les ET, son espèce soeur, D. simulans, prend de plus en plus d’importance grâce à son génome qui d’une part porte moins d’ET que D. melanogaster et d’autre part présente une forte variation du nombre de copies d’ET entre populations. Nous nous sommes attachés à comprendre les mécanismes responsables de ces différences en étudiant un grand nombre d’ET dans des échantillons de populations récoltés sur l’ensemble du globe. Nos résultats sont en faveur d’une invasion récente du génome de D. simulans par la plupart de ses ET (Biémont et al., 2003 ; Vieira et Biémont, 2004). Cela n’exclut pas bien sûr l’alternative de pertes de certains ET chez cette espèce, ou bien l’existence de caractéristiques particulières qui lui permettraient de résister à l’invasion. L’analyse des copies des ET du génome séquencé de D. melanogaster montre un fort turn-over de copies, ce qui confirme l’intense activité dans ce génome (Lerat et al., 2002a, 2002b, 2003). On attend une version utilisable du génome de D. simulans pour analyser ses copies et avoir ainsi une comparaison directe avec D. melanogaster.
L’analyse détaillée des séquences du rétrotransposon 412 et de l’expression de cet élément dans les populations naturelles des deux espèces a ainsi montré le rôle important joué par la recombinaison dans l’émergence de nouveaux éléments qui pourraient alors acquérir de nouvelles capacités d’invasion (Mugnier et al., 2005), et l’apparition de mécanismes de régulation de l’expression de l’élément 412 dont l’intensité varie selon les populations et le nombre de copies (Borie et al., 2002). Ce dernier résultat montre l’importance des études de populations dans l’analyse des transcriptomes. L’expression des ET est non seulement tissu-spécifique mais dépend des populations, ce qui est en accord avec les récents travaux montrant que l’expression des gènes varie entre populations (Whitehead and Crawford, 2005, Genome Biology) et que de nombreux facteurs épigénétiques, comme la nutrition, interviennent sur la régulations de l’expression des ET (Waterland et al., 2005, Nutrition). Les modifications de l’environnement seraient alors un puissant facteur de changements dans la composition et la structuration des génomes. Ceci peut rendre compte de la variation de taille des génomes (estimée par cytométrie en flux) entre les populations ancestrales, africaines et les populations dérivées de Drosophila melanogaster (Vieira et al., 2002 ; Nardon et al., 2003) et révèle le rôle important que peut jouer la colonisation dans les changements de composition des génomes, même si le temps nécessaire est sans doute assez long (Nardon et al., 2005). Une étude détaillée des espèces du sous-groupe melanogaster a mis en valeur les variations de taille des génomes au cours de l’évolution avec des gains et pertes non seulement d’éléments transposables mais aussi d’autres séquences répétées, telles les séquences satellites, dont le rôle dans l’évolution des génomes a sans doute été trop longtemps négligé.
La découverte de l’élément tirant, qui n’est représenté que dans quelques populations de D. simulans, nous ouvre des perspectives importantes pour la compréhension des phases d’invasion des génomes par les ETs. Ainsi un scénario possible est que certaines copies de tirant auraient envahi le génome de l’ancêtre de D. melanogaster et D. simulans, puis auraient été perdues chez D. melanogaster lors de sa migration hors d’Afrique. La migration de D. simulans aurait par contre concerné des mouches dénuées de copies de tirant actives qu’on trouve encore cependant dans quelques populations africaines (thèse de M. Fablet).
Ces données montrent que la compréhension des génomes nécessite une approche pluridisciplinaire, et qu’il faut intégrer les données de la biologie moléculaire à celles de la génétique des populations et de l’écologie des espèces, et les associer aux résultats de l’analyse bioinformatique des génomes séquencés. Cette dernière approche, bien qu’elle ne concerne que le génome d’un individu et ne renseigne pas sur le polymorphisme, est riche d’enseignements car ces génomes portent les marques de leur histoire évolutive. C’est ainsi que l’analyse bioinformatique des génomes séquencés de la drosophile et du nématode nous a permis, en collaboration avec L. Ségalat (CGMC, Lyon) et L. Duret, M. Gouy et G. Marais, (équipe BGE) de préciser la part jouée par les recombinaisons, la sélection et le choix des séquences d’intégration dans la distribution des copies d’ET sur les chromosomes (Rizzon et al., 2002, 2003), ce qui a donné une suite intéressante aux travaux initiaux de C Hoogland et al., de 1996 (Genetics, 1996). Enfin une approche de modélisation mathématique de l’invasion des ET dans les populations naturelles à partir de modèles classiques fondés sur le taux de transposition, la sélection et la migration, a permis de se faire une idée des dynamiques en cours dès les premières générations et a fourni une explication possible à de nombreuses données observées dans les populations naturelles (Deceliere et al., 2005, en collaboration avec S. Charles, équipe Baobab) (Figure 2).
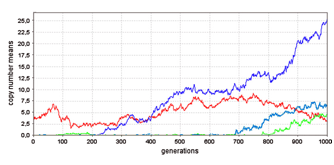
Figure 2 : Illustration du rôle important de la migration entre populations dans la dynamique des éléments transposables des génomes. Simulation de l’évolution du nombre de copies d’un élément transposable dans quatre populations au cours des générations. Taux de migration entre populations, m = 0.02. Taille des populations: 50 individus. Taux de transposition: 10-4. Seule la population rouge avait 4 copies au début de la simulation. Travail de thèse de G. Decelière (collaboration S. Charles, Y. Letrillard).
2) Perspectives
Nos connaissances acquises grâce au tandem D. melanogaster/D. simulans, nous amènent maintenant à une comparaison d’espèces pour rechercher les mécanismes ayant conduit à des modifications drastiques de la composition, de la structure et donc de la taille des génomes entre espèces. Outre les espèces du genre Drosophila et plus particulièrement du sous-groupe melanogaster, nous développerons une recherche sur les moustiques et plus particulièrement Anopheles gambiae, le vecteur de la Malaria (collaboration : D. Fontenille, IRD de Montpellier). Cela nous permettra une comparaison de deux génomes d’insectes qui ont eu des évolutions divergentes et diffèrent pour leur écologie. Ces travaux seront fondés sur les outils moléculaires et cytologiques de manière à aborder les questions de génomique fonctionnelle, et donc les mécanismes fins de l’évolution, au niveau des populations.
L’analyse bioinformatique sera renforcée par la connaissance du génome de l’espèce D. pseudoobscura puis par celle des génomes des autres espèces du sous-groupe melanogaster avec une mention particulière pour D. simulans, dont une première version du génome vient d’être publiée et pour laquelle plusieurs individus seront séquencés.
Notre objectif est d’accentuer les interactions entre biologie moléculaire, bioinformatique et génétique des populations en confrontant les données populationnelles à celles des génomes séquencés. C’est de cette confrontation que naîtra la complémentarité indispensable à une connaissance globale des mécanismes qui déterminent la structure et la composition des génomes et régulent l’expression de leurs composants.
BioInformatique et Génomique Évolutive
Le principal domaine de recherche de l’équipe est l’évolution moléculaire à l’aide d’analyses bioinformatiques des séquences nucléotidiques. Ces dernières années, les travaux à l’échelle génomique entière sont devenus prépondérants avec la publication du séquençage génomique complet de plusieurs dizaines d’organismes. L’approche comparative est un outil majeur de cette recherche car elle permet d’analyser le processus évolutif en détail. En soutien à cette thématique de recherche, des développements bioinformatiques importants ont été réalisés, notamment au plan des banques de familles de séquences homologues, et de services bioinformatiques ouverts à la communauté. Les principaux résultats obtenus sont brièvement présentés dans ce texte. En particulier une avancée majeure a été obtenue au sujet de l’évolution des génomes de vertébrés : l’élucidation du mécanisme à l’origine de la création et du maintien des isochores, en lien avec le phénomène général qu’est la recombinaison.
1) Influence de la recombinaison sur l’évolution des génomes de métazoaires : de l’origine des isochores à la dégénérescence des chromosomes sexuels
(L. Duret, G. Marais, D. Mouchiroud, A. Khelifi, J. Meunier)
Le long des chromosomes de mammifères, il existe une forte variabilité de la composition en bases G+C, sur une échelle de centaines de kilobases. Cette structuration compositionnelle appelée, ‘isochore’, est fortement corrélée avec d’autres caractéristiques importantes de l’organisation du génome: densité en gènes, taille des introns, distribution des éléments transposables, taux de recombinaison, bandes chromosomiques et réplication. Ainsi, la structure en isochores est une caractéristique fondamentale de l’organisation du génome des mammifères. Pour comprendre la signification de cette organisation, il est donc important de déterminer si cette structure en isochores est le résultat de l’action de la sélection, ou bien la conséquence de processus évolutifs neutres. Cette question à laquelle notre équipe s’intéresse depuis plus de quinze ans a fait l’objet de nombreux débats dans la communauté scientifique (pour revue, voir Eyre-Walker et Hurst 2001, Nature Rev Genet 2:549).
Grâce à l’accumulation de séquences génomiques chez différents mammifères, et notamment chez des primates (homme, chimpanzé, babouin), nous avons pu analyser en détail les variations des patrons de substitution le long de notre génome (thèse J. Meunier). Nous avons ainsi pu démontrer que la recombinaison a un impact majeur sur l’évolution du taux de G+C (Meunier et Duret 2004, article sélectionné par « Faculty of 1000 » : voir). Cet effet est dû à un processus évolutif neutre: la conversion génique biaisée (BGC) (Duret et al., 2002). Des variations rapides des taux de recombinaison au cours de l’évolution, liées à des modifications des caryotypes, sont très certainement à l’origine de l’apparition des isochores GC-riches chez l’ancêtre commun des amniotes (mammifères, reptiles, oiseaux) et de leur disparition progressive chez les mammifères (Duret et al., 2002 ; Marais et Galtier, 2003 ; Belle et al., 2004 ; Meunier et Duret, 2004). L’impact de la conversion génique biaisée sur l’évolution de la composition en base G+C a également été mis en évidence par l’étude du patron de substitutions des rétropseudogènes humains et murins en fonction de leur contexte d’insertion et notamment le taux de recombinaison local (Thèse Adel Khelifi). Nous avons par ailleurs montré que chez les invertébrés, il existe également une corrélation entre recombinaison et composition en bases (Marais et al. 2003). En fait, il est vraisemblable que le processus de BGC affecte tous les organismes sexués (Duret, 2002 ; Marais, 2003 ; Marais et al., 2004) .
Ces différents résultats sont importants, non seulement pour la compréhension de l’origine et de l’évolution des isochores, mais aussi plus généralement pour l’interprétation des processus de substitution. Jusqu’à présent, ces processus étaient considérés comme résultant essentiellement de la sélection et de la dérive. Nos résultats indiquent qu’en plus de ces facteurs, la conversion génique biaisée (et donc la recombinaison) joue un rôle majeur dans l’évolution des génomes des organismes sexués.
Cette découverte nous a amené à revisiter les relations entre recombinaison et évolution des génomes (Thèse Gabriel Marais). En effet, à l’origine, les biologistes évolutionistes se sont intéressés à la recombinaison pour essayer de comprendre les raisons pour lesquelles la reproduction sexuée est apparue au cours de l’évolution. Il a notamment été démontré que la recombinaison permet d’augmenter l’efficacité de la sélection naturelle (effet Hill-Robertson). C’est pourquoi de nombreux auteurs interprétaient les corrélations entre recombinaison et divers paramètres génomiques (taille des introns, répartition des éléments transposables, usage des codons, ...) comme des conséquences de cet effet Hill-Robertson. Nous avons montré qu’en réalité, beaucoup de ces corrélations résultent des effets non-sélectifs de la recombinaison (Rizzon et al., 2002, 2003 ; Marais et Piganeau, 2002 ; Marais et al., 2003). Cela ne signifie pas que la recombinaison ne procure aucun avantage sélectif. En particulier, l’analyse de l’évolution des chromosomes sexuels de plantes dioïques montre que tout comme chez les mammifères, la suppression de recombinaison s’accompagne d’une dégénérescence des gènes localisés sur le chromosome Y (Nicolas et al., 2005). Pour comprendre l’impact de la recombinaison (et donc de la reproduction sexuée) sur l’évolution des génomes, il est donc essentiel de prendre en compte à la fois les processus sélectifs et neutres affectés par la recombinaison.
Il faut souligner que ces travaux sur l’analyse de l’évolution des génomes sont menés en collaboration avec plusieurs équipes du département. Notamment l’étude de la relation entre recombinaison et insertion d’éléments transposable est un travail conjoint avec l’équipe Génomes et Populations (Rizzon et al., 2002, 2003). Par ailleurs, l’équipe Baobab s’intéresse également à l’évolution et à la structure des génomes et a notamment développé de nouvelles méthodes et bases de données pour la détection des isochores et pour l’analyse des patrons d’évolution le long des génomes (cf ci-dessous V- Baobab 2) et 3)).
2) Étudier l’évolution des génomes pour mieux comprendre leur fonctionnement
(L. Duret, D. Mouchiroud, A. Khelifi , J. Meunier, M. Sémon)
L’étude de l’évolution des génomes permet également de mettre en évidence certaines caractéristiques de leur fonctionnement. Par exemple, en analysant les patrons de substitution sur les dinucléotides CpG, nous avons pu démontrer qu’il existe chez les primates un mécanisme homologie-dépendant conduisant à l’hyperméthylation des séquences répétées (éléments transposables ou pseudogènes) (Meunier et al., 2005). Ce résultat est important car il est la première démonstration directe de l’existence d’un mécanisme de défense contre l’invasion des éléments transposables dans des génomes de mammifères. Cet article a été sélectionné par « Faculty of 1000 » (voir).
Par ailleurs, nous avons montré que les éléments transposables LINE et HERV sont sous-représentés dans les introns, notamment dans l’orientation correspondant au sens de la transcription, vraisemblablement parce que leurs éléments régulateurs perturbent l’expression des gènes dans lesquels ils s’insèrent. Nous avons tiré parti de cette observation pour développer une méthode de prédiction des régions transcrites dans le génome humain, basée sur l’analyse de la distribution des éléments transposables. Nous avons ainsi montré que les unités de transcription fonctionnelles couvrent au moins 50% de notre génome, et qu’un tiers d’entre elles correspondent à des gènes non-codants (Sémon et Duret, 2004).
3) Processus évolutifs structurant les génomes bactériens
(J. Lobry)
Un des objectifs de notre démarche est d’identifier les processus évolutifs à l’origine de la structure des génomes actuels, particulièrement bactériens, et d’étudier la relation entre cette structuration et le fonctionnement des génomes.
La figure 3 donne un résultat illustrant cet objectif. Ce résultat (Lobry, 2004) est très novateur puisqu’il associe pour la première fois un phénomène sélectif (le coût métabolique de la synthèse protéique) à un phénomène génomique majeur, la composition en bases G+C. Or, depuis 40 ans toutes les hypothèses conférant une valeur adaptative au contenu génomique bactérien en G+C, particulièrement en relation avec la température optimale de croissance, avaient été infirmées.
Le taux de G+C des génomes bactériens varie de 25 % à 75 %, et on sait depuis 2002 (Naya et al., 2002 JMolEvol 55 :260) que le taux de G+C des bactéries aérobies est significativement plus élevé que celui des bactéries anaérobies (graphique du bas).
Le coût métabolique en aérobiose de synthèse d’une protéine dépend de sa composition en acides aminés, et les protéines fortement exprimées évitent d’utiliser des acides aminés trop coûteux, la différence observée entre une protéine faiblement et fortement exprimée est de l’ordre de 2 équivalents ATP par acide aminé en moyenne (barre verticale en haut à droite). On sait par ailleurs depuis longtemps que le taux de G+C a une forte influence sur la composition en acides aminés des protéines. Le graphique du haut montre que cette influence va dans le bon sens puisque les bactéries aérobies, riches en G+C, ont des protéomes sensiblement moins coûteux. On a ainsi ici un exemple de structure génomique forte, le taux de G+C des génomes bactériens, mise en relation avec un mécanisme, la biosynthèse des protéines, suggérant un scénario évolutif par exaptation à l’origine de la variabilité observée.
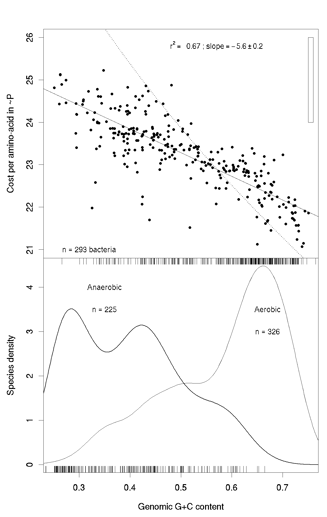
Figure 3 : Relation entre le taux de G+C des génomes bactériens et le coût métabolique de l’expression des protéines codées par ces génomes (voir texte pour plus d’explications).
4) Génomique comparative bactérienne
(M. Gouy, G. Perrière, V. Daubin, A. Calteau)
L’activité du laboratoire en génomique comparative bactérienne s’articule autour de deux axes interconnectés, l’un biologique, concernant l’étude de transferts interspécifiques de gènes, l’autre méthodologique regardant plus particulièrement le développement d’approches phylogénétiques.
Les transferts de gènes ont un impact majeur à la fois sur la biologie des organismes et sur notre capacité à reconstruire leur histoire et comprendre leur évolution. Dans le cadre de leurs thèses respectives, Vincent Daubin (thèse 2002) et Alexandra Calteau (thèse en cours) ont analysé les différentes approches possibles pour explorer la phylogénie des bactéries, et leur robustesse aux transferts de gènes. Ils ont proposé des approches originales pour la détection de ces échanges et l’inférence phylogénétique basée sur les grands jeux de données (typiquement, en utilisant des centaines de gènes). Notamment, ces travaux ont donné lieu aux premières applications de méthodes dites de "superarbre" pour l’inférence d’une phylogénie universelle du vivant et ont permis a la fois de mettre en évidence un ensemble de gènes relativement robustes aux transferts et d’exploiter l’ensemble de ces gènes en tant que marqueurs phylogénétiques (Daubin et al., 2002 ; Calteau et al., 2004).
En parallèle, nos travaux ont porté sur l’étude de l’impact des transferts récents sur les génomes et leur rôle biologique. Dans un premier temps, ils ont permis de mieux comprendre la nature des gènes les plus susceptibles d’être transférés. Il apparaît que contrairement à l’idée la plus répandue, la majorité de ces gènes n’est pas transmise directement d’une bactérie à une autre, mais séjourne vraisemblablement pour de longues périodes dans des vecteurs tels que les bactériophages. Cette découverte suggère que ces gènes contribuent généralement d’abord au succès reproducteur du bactériophage, et seulement secondairement à celui de la bactérie réceptrice (Daubin et al., 2003 ; Daubin et Ochman, 2004a ; Daubin et Ochman, 2004b). D’autre part, des recherches ont été menées dans le but d’éclaircir les relations existant entre bactéries et archées hyperthermophiles, et plus particulièrement le rôle des transferts horizontaux de gènes dans l’adaptation à la vie à haute température. La quantification des événements de transferts horizontaux d’archées chez quatre bactéries hyperthermophiles et thermophiles a démontré que les premières ont subi statistiquement plus de transferts horizontaux. La majorité de ces transferts implique des gènes codant pour des protéines de fonction inconnue. Ces observations suggèrent un rôle potentiel de ces protéines inconnues dans l’adaptation à l’hyperthermophilie (Calteau et al., 2005).
5) Étude phylogénétique du génome de la microsporidie Encephalitoozoon cuniculi
(M. Gouy, F. Thomarat)
En collaboration avec l’instigateur (Christian Vivarès, Université de Clermont-Ferrand) du séquençage génomique de cet organisme, l’analyse phylogénétique de ce génome a conduit à deux nouvelles publications. La présence d’un organite, le mitosome, dérivé de la mitochondrie, en analogie avec d’autres protistes ‘amitochondriaux’ comme Entamoeba, et consacré à l’assemblage des clusters Fe-S, fonction mitochondriale essentielle chez tous les eucaryotes, a été prédite (Vivares et al., 2002), prédiction confirmée expérimentalement par un laboratoire anglais (Williams et al., 2002 Nature 418 :865). La parenté évolutive entre microsporidies et champignons a été étayée à l’échelle du génome entier (Thomarat et al., 2004). Cette analyse montre aussi l’effet de la vitesse évolutive relative de chaque protéine sur les reconstructions phylogénétiques et documente la présence de l’artefact « d’attraction des longues branches » sur des données réelles.
6) Banques de données et services en ligne
(M. Gouy, G. Perrière, L. Duret, J. Thioulouse, A. Khelifi, J. Grassot, JF Dufayard)
Afin de faciliter les analyses de génomique comparative dans lesquelles notre équipe est impliquée, nous sommes intervenus dans le développement de plusieurs banques de données de séquences homologues.
Historiquement, la première d’entre elles fut HOVERGEN (Homologous Vertebrates Genes Database), consacrée aux gènes protéiques de vertébrés (Duret et al., 1994). Devant le succès remporté par ce système, nous avons alors décidé de mettre en place HOBACGEN (Homologous Bacterial Genes Database), dédiée aux gènes de bactéries, d’archées et de levure (Perrière et al., 2000). Parmi les avancées d’HOBACGEN figuraient une suppression de la redondance dans les séquences, le développement d’une interface spécialisée portable, et la mise en place d’une structure de type client-serveur. Fut ensuite développée HOGENOM (Homologous Sequences in Complete Genomes), dédiée à l’ensemble des génomes complètement séquencés. Par ailleurs, des systèmes plus spécialisés furent mis en place : NuReBase (Nuclear Receptors Database), consacrée aux gènes de récepteurs nucléaires (Ruau et al., 2004), RTKdb (Receptor Tyrosine Kinase Database) pour les gènes de récepteurs à Tyrosine Kinase (Grassot et al., 2003), et HOPPSIGEN, pour les séquences de rétropseudogènes (Khelifi et al., 2005).
L’organisation sous la forme d’une structure de type client/serveur nous a permis de supprimer les problèmes d’installation et de mise à jour précédemment rencontrés avec la première version d’HOVERGEN. Côté client un programme Java, FamFetch (Perrière et al., 2000) permet de visualiser séquences, alignements et arbres par l’intermédiaire d’une interface graphique. FamFetch permet d’effectuer des sélections sur les familles en utilisant plusieurs critères : mots clés, liste de taxons, noms ou numéros d’accession des séquences, nombre de taxons différents représentés, etc. Une fonctionnalité récemment ajoutée permet également de composer des requêtes portant sur la topologie des arbres figurant dans les banques (Dufayard et al., 2005). Ainsi, des motifs simples ou complexes – impliquant par exemple la présence de duplications dans une famille – peuvent être rentrés afin de récupérer les familles correspondantes. Il s’agit donc d’un outil particulièrement efficace pour identifier de manière automatique les orthologues présents à l’intérieur de familles de gènes.
Une part importante de nos développements informatiques passent dans la mise en place et la maintenance de services au niveau du serveur Web du Pôle Bioinformatique Lyonnais (PBIL, composante du PRABI, Pôle Rhône-Alpin de Bioinformatique (Perrière et al., 2003)). Ce serveur permet bien sûr d’accéder aux banques de séquences homologues décrites ci-dessus, mais également à tout un ensemble de banques de données publiques, couramment utilisées en bioinformatique : GenBank, EMBL, SWISS-PROT, RefSeq, etc. Parmi les caractéristiques les plus notables de ce serveur figurent la possibilité de visualiser les alignements et les arbres phylogénétiques intégrés dans nos banques de familles. Les autres services proposés par le PBIL consistent en des programmes de recherche de similarité, d’alignement, ou d’analyse statistique des séquences. Pour exemple, un logiciel de prédiction de promoteur à ilôts CpG a été proposé dans les génomes complets de mammifères (CpGProd) (Ponger et Mouchiroud, 2002). De plus, une version « en ligne » du logiciel R est disponible, version intégrant la totalité des bibliothèques agréées par le CRAN. Parmi ces bibliothèques figure seqinR, développée par notre équipe, et permettant d’effectuer des analyses statistiques complexes sur les séquences des banques (Charif et al., 2005).
7) Perspectives
Nos projets de recherche se situent sur deux axes principaux : l’analyse des processus d’évolution des génomes et le développement d’outils bioinformatiques pour la génomique comparative. L’étude des processus évolutifs est aujourd’hui révolutionnée par le changement d’échelle du volume des données disponibles. Ainsi, le projet d’analyse systématique des génomes d’espèces proches détaillé ici, illustre bien comment le niveau génomique devrait permettre de faire avancer significativement les connaissances sur les processus évolutifs. Au plan de la phylogénie moléculaire, nous poursuivrons les analyses à l’échelle génomique procaryote, et espérons pouvoir grandement améliorer la résolution obtenue et l’identification des transferts horizontaux. Par ailleurs, plusieurs projets méthodologiques sont prévus, en particulier autour de l’analyse des données de puces à ADN et de la biodiversité microbienne. De plus, la venue de Daniel Kahn sera l’occasion de développer de nouveaux projets sur l’étude de l’évolution de la modularité des protéines et de migrer la base ProDom de familles de domaines protéiques (Bru et al., 2005) au laboratoire (cf. dossier D. Kahn). Cette migration devrait s’accompagner de son intégration avec les bases de données de gènes homologues développées au PBIL (HOGENOM, HOVERGEN).
Génomique micro-évolutive : élucider les processus évolutifs et révéler les contraintes fonctionnelles dans les génomes complets
Pour analyser précisément l’évolution des séquences génomiques, deux conditions sont requises. Tout d’abord, pour pouvoir detecter l’ensemble des évènements évolutifs, il est nécessaire de comparer des espèces suffisamment proches. En effet, si les changements sont trop nombreux, alors il devient impossible de reconstituer la succession des évènements évolutifs ayant conduit aux génomes contemporains (on parle de saturation du signal phylogénétique). Par ailleurs, pour pouvoir orienter ces évènements évolutifs il est nécessaire de disposer des génomes d’au moins trois espèces différentes dont la phylogénie est connue.
Jusqu’à présent, la plupart des génomes séquencés provenait d’espèces relativement distantes entre elles. De ce fait, il n’était possible d’analyser les processus évolutifs que sur les régions du génome qui évoluent le plus lentement (typiquement les régions codantes des gènes protéiques). Ainsi, très peu de choses sont connues sur l’évolution des séquences non-codantes (intron, régions intergéniques) qui constituent pourtant la fraction principale de nombreux génomes eucaryotes. Cette situation est en train de changer, grâce à plusieurs projets de séquençage d’espèces proches dans divers groupes taxonomiques (primates, drosophiles, levures, bactéries, archées). Il faut noter également que plusieurs projets ont également été mis en place pour analyser le polymorphisme à l’échelle de génomes complets, non seulement pour les populations humaines, mais également pour divers organismes modèles.
Ainsi, un de nos principaux objectifs dans les années qui viennent est de tirer parti des ces données de séquence et de polymorphisme pour déterminer précisément les processus évolutifs sur l’intégralité d’un génome. Nous nous intéressons non-seulement aux changements ponctuels (substitutions, petites insertions ou délétions), mais également aux insertions d’éléments transposables et aux remaniements génomiques à plus grande échelle. Notre projet consiste à utiliser cette approche de génomique comparative à petite échelle évolutive pour investiguer différentes questions : la détection et la quantification de la sélection dans le génome, l’impact de la recombinaison sur l’évolution des génomes, les relations entre évolution de l’expression et évolution du génome et notamment l’impact des éléments transposables, l’évolution des gènes d’ARN non codant et des régions régulatrices.
Ce projet implique des développements bioinformatiques importants. Nous allons établir une procédure pour générer des alignements multiples de génomes complets: détection des régions génomiques orthologues, reconstruction des séquences ancestrales et inférences des réarrangements chromosomiques. Nous allons également développer une base de données permettant de gérer ces alignements et les informations sur les évènements évolutifs inférés. Les alignements génomiques multiples seront réalisés sur des génomes de primates (homme, chimpanzé, babouin), de drosophiles (D. melanogaster, D. simulans et D. yakuba) et de divers micro-organismes.
Évolution des répertoires de gènes
L’évolution du répertoire de gènes joue un rôle fondamental dans l’adaptation des espèces à leur environnement. La comparaison de génomes de différentes souches bactériennes a démontré que cette évolution pouvait être extraordinairement rapide. Par exemple, les trois souches d’Escherichia coli MG1655, 0157:H7 et CFT073 possèdent respectivement 4289, 5060 et 5016 gènes, parmi lesquels seulement 2996 sont partagés. Chaque souche possède entre 585 et 1623 gènes qui lui sont spécifiques (Welch et al., 2002 PNAS 99:17020). Beaucoup de ces gènes orphelins ont été acquis par transfert horizontal (Daubin et al., 2003; Daubin et Ochman, 2004). Chez les eucaryotes, les transferts horizontaux de gènes semblent moins fréquents que chez les bactéries, mais il existe également d’autres mécanismes qui contribuent à l’évolution du répertoire génique : duplication de gènes, de segments chromosomiques, voire de génomes entiers ; perte de gènes ; formation de nouveaux gènes par réarrangement entre gènes pré-existants.
Nous comptons tout d’abord analyser l’évolution des répertoires de gènes au sein des mammifères : quels sont les gènes nouveaux apparus depuis la divergence entre rongeurs et primates ? Comment se sont-ils formés ? Quels gènes ont été dupliqués dans chacune des deux lignées ? Quels gènes ont été perdus ? Pour répondre à ces questions, il est nécessaire d’établir l’inventaire exhaustif des gènes orthologues entre l’homme et la souris. Curieusement, alors qu’il est couramment admis que les répertoires géniques de ces deux espèces sont quasiment identiques, il y a plus de 30 % des gènes annotés dans le génome humain qui n’ont aucun orthologue identifié chez la souris (données Ensembl, septembre 2005). Jusqu’à présent, cette identification d’orthologues était effectuée essentiellement sur la base de recherche de similarité entre séquences (méthode dite des « meilleurs hits réciproques »), dont on sait qu’elle présente de nombreuses limitations, notamment lorsque les gènes ont subi des duplications. Nous allons donc développer une nouvelle procédure d’identification d’orthologues basée à la fois sur l’analyse phylogénétique des familles de gènes (et pour cela nous utiliserons les outils que nous avons déjà développés pour HOVERGEN) et sur l’analyse de la conservation de l’ordre des gènes le long des génomes de mammifères. Notre objectif est d’obtenir un inventaire réellement exhaustif des gènes orthologues au sein des mammifères. Nous exploiterons l’ensemble des génomes de mammifères séquencés à ce jour pour déterminer si les gènes humains qui n’ont pas d’orthologues chez la souris correspondent à des gènes qui ont été perdus chez les rongeurs, ou bien acquis chez les primates. Ce travail d’inventaire systématique des gènes orthologues représente un enjeu considérable, non seulement pour la génomique évolutive, mais aussi pour l’analyse fonctionnelle des génomes.
Par ailleurs nous allons analyser l’évolution du répertoire de gènes chez Paramecium tetraurelia. Le génome de cette paramécie vient d’être séquencé par le Génoscope et nous participons actuellement à son analyse, en collaboration avec les laboratoires de Jean Cohen (Gif-sur-Yvette) et Eric Meyer (ENS-Ulm). Cet eucaryote unicellulaire possède un génome de 70 Mb, extrêmement dense en gènes (près de 40,000 gènes). Ce nombre très élevé de gènes est la conséquence d’au moins deux évènements de duplication complète du génome au cours de l’évolution des paramécies. De tels événements de polyploïdisation sont relativement rares mais peuvent avoir un impact majeur sur évolution des espèces. En effet, les duplications de gènes jouent un rôle fondamental dans l’évolution de nouvelles fonctions et donc dans l’adaptation des espèces. La séquence du génome de la paramécie présente un intérêt considérable pour la compréhension de ce phénomène. En effet, les duplications génomiques mises en évidence jusqu’à présent dans les génomes séquencés (levures, plantes et poissons) sont relativement anciennes, et la grande majorité des gènes dupliqués a été perdue. A contrario, chez la paramécie, seulement 33 % des gènes ont été perdus depuis la dernière duplication, et le génome a subi très peu de remaniements chromosomiques. Il est donc aisé d’établir le caryotype ancestral et de reconstituer les scénarios évolutifs pour chacune des deux duplications. Ainsi, ce génome de la paramécie nous permet pour la première fois d’analyser directement les conséquences d’une duplication génomique sur l’évolution de la fonction des gènes : quels gènes sont perdus après duplication ? quels sont ceux qui sont retenus ? comment évolue le patron d’expression des gènes dupliqués ?
Analyse des données de puces à ADN et de biodiversité microbienne
Notre objectif est ici de croiser les développements déjà existants dans les domaines biologique et statistique, pour aboutir à une meilleure collaboration et à de nouveaux résultats. Sur le plan biologique, deux types de travaux sont concernés : des travaux appliqués au traitement du cancer, et des travaux se rattachant à l’écologie microbienne. Les travaux sur le cancer concernent en particulier la comparaison des résultats obtenus avec différents types de puces, l’analyse de la réponse à différents types de traitements, la prévision du pronostic pour des patients atteints de divers types de cancer, et la correction de l’optimisme de la prédiction des modèles pronostiques (en collaboration avec les équipes du départment « BioMaths-Santé »). Les travaux se rattachant à l’écologie microbienne visent à évaluer la diversité bactérienne dans des milieux complexes comme le sol, avec des objectifs de type génotypage, analyse phylogénique, et taxonomie. Sur le plan statistique, nos approches sont de type analyse statistique multivariée (en particulier analyse en composantes principales, analyse discriminante, analyse de co-inertie, classification automatique), méthodes de re-échantillonnage (tests de Monte-Carlo, tests de permutation), modélisation, et statistique Bayesienne. Notre objectif est donc de renouveler l’approche statistique des données de puces à ADN, qui est actuellement abordée presque uniquement selon l’angle univarié et au moyen de la statistique inférentielle classique (analyse de variance gène par gène dans le cas des données d’expression), afin d’améliorer leurs capacités de diagnostic et de prévision.
Étude de la modularité des protéines
Étude de l’évolution de la modularité des protéines par une approche Bayésienne
Lors d’une précédente étude, nous avons utilisé les réseaux Bayésiens pour reconstituer les scénarios les plus probables d’évolution des familles de domaines protéiques. Nous étudierons la possibilité d’incorporer dans ce modèle Bayésien l’information phylogénétique contenue dans les séquences. La même méthodologie sera utilisée pour reconstituer les scénarios d’évolution des familles de protéines modulaires. Il sera alors possible de croiser ces scénarios pour reconstituer des scénarios évolutifs complexes qui prennent en compte l’apparition de nouveaux arrangements en domaines, débouchant sur une « histoire » de la modularité des protéines : à quel moment est apparu un arrangement en domaines particulier ? À partir de quelles espèces ancestrales ? Dans quels phyla cette invention moléculaire s’est-elle stabilisée ? Cette dimension évolutive de l’analyse des arrangements en domaines pourrait être précieuse en vue d’une interprétation fonctionnelle des séquences.
Utilisation de la décomposition en domaines des protéines pour l’analyse de l’interactome
Il est maintenant bien établi que de nombreuses interactions entre protéines peuvent en première approximation être expliquées par des interactions entre domaines, parfois spécialisés. La combinatoire des interactions entre protéines résulte donc d’une part de la combinatoire des arrangements en domaines, d’autre part de la spécificité de reconnaissance.
Nous proposons d’exploiter la combinatoire des domaines observés dans les paires de protéines en interaction, afin d’identifier les couples de domaines sur-représentés. On associera donc à chaque couple de domaines une probabilité d’interaction. Réciproquement, ces probabilités serviront à calculer des probabilités d’interaction entre protéines, sur la base de leurs arrangements en domaines. De la sorte on pourra prédire dans un génome un ensemble d’interactions plausibles, triées par probabilité décroissante.
Les probabilités d’interactions entre domaines seront intégrées à ProDom , ce qui fournira l’ensemble des domaines susceptibles d’interagir avec un domaine donné. Nous diffuserons également un outil d’estimation de la probabilité d’interaction de deux protéines sur la base de leurs arrangements en domaines. Ceci permettra en particulier d’identifier, à l’intérieur d’un génome, des protéines susceptibles d’interagir avec une protéine donnée.
Développements bioinformatiques
Intégration de ProDom avec les bases du PRABI
Il existe deux points de vue principaux dans l’analyse des familles protéiques : une analyse basée sur l’homologie de séquences entières, et une analyse basée sur les familles de domaines. Le premier point de vue est pleinement exploité dans les bases HOGENOM, HOBACGEN et HOVERGEN. Le second point de vue considère les protéines comme des arrangements combinatoires de domaines, et est à la base de ProDom. Ces deux poins de vue complémentaires méritent d’être croisés, à la fois dans l’optique de l’annotation des séquences et dans celle de l’évolution des protéines. En conséquence nous viserons à intégrer les deux points de vue dans l’environnement du PBIL, par exemple en identifiant les familles d’HOGENOM à l’intérieur des familles de ProDom.
Déploiement de ProDom sur la grille informatique
Un deuxième axe de travail concerne l’intégration de ProDom sur la grille informatique, dans le cadre d’un Réseau d’excellence financé par le FP6 (REX « EMBRACE », coordonné par Graham Cameron, EBI). Il s’agit d’un projet technologique qui vise à intégrer les bases de données biologiques et à rendre possibles des requêtes utilisant plusieurs bases de données sur la grille. Les standards technologiques seront définis dans le cadre d’un work package coordonné par Vincent Breton (CNRS, Clermont-Ferrand). Pour notre part nous développerons des APIs spécifiques à ProDom conformes à ces standards. Elles permettront à d’autres programmes d’accéder aux différentes fonctionnalités de ProDom à travers la grille : alignements multiples, arbres phylogénétiques, représentation graphique des arrangements en domaines, représentation structurale, recherche d’homologies, requêtes sur les arrangements en domaines. Le résultat sera la possibilité de réaliser des opérations complexes impliquant à la fois ProDom et d’autres bases de données au travers de la grille.
Baobab
1) Introduction
L’équipe BAOBAB appartient à deux structures de recherche différentes, le projet (équipe) HELIX de l’INRIA bi-localisé à Grenoble et à Lyon et le Laboratoire de Biométrie et Biologie Évolutive (LBBE) de l’Université Claude Bernard, Lyon I. Ce dernier est composé essentiellement de biologistes, bio-mathématiciens et bio-informaticiens alors que le projet HELIX est composé en majorité d’informaticiens et de bio-informaticiens.
Une telle multidisciplinarité se retrouve de façon sans doute encore plus prononcée au sein de l’équipe BAOBAB. Certains de ses membres ont ainsi suivi un cursus universitaire en mathématiques (principalement combinatoire), statistiques ou informatique (théorique) jusqu’à leur thèse, ne développant un intérêt pour la biologie qu’à ce niveau, ou même plus tard (lors d’un post-doctorat). L’équipe intègre également des membres ayant suivi jusqu’à la thèse un cursus en biologie (biologie moléculaire, évolution, biochimie) et qui ont été séduits par l’idée d’utiliser les mathématiques et/ou l’informatique afin de répondre à des questions biologiques de portées diverses. Cette idée, et les difficultés à interpréter les résultats obtenus avec les modèles mathématiques et les algorithmes existants, les ont encouragés à s’intéresser de plus près à ces modèles et algorithmes comme un sujet de recherche en soi.
L’équipe possède ainsi des objectifs qui reflètent à la fois ses origines diverses et sa passion commune pour la biologie. Ces objectifs vont de :
- identifier les problèmes biologiques qui sont maintenant, ou seront dans les prochaines années importants afin d’améliorer notre connaissance des systèmes biologiques et qui peuvent être traités par des méthodes formelles, en passant par
- obtenir des modèles mathématiques bien définis et de plus en plus précis de ces problèmes, jusqu’à
- développer les algorithmes statistiques et combinatoires qui vont permettre d’apporter une solution au moins partielle à ces problèmes.
Le dernier point implique également de développer une recherche en mathématiques et en informatique théorique « pures ». Outre une question d’intérêt propre de certains membres de l’équipe, un tel développement est absolument indispensable si l’on espère pouvoir traiter de manière satisfaisante des problèmes dont la nature est souvent très complexe et hautement combinatoire. De la même façon, la passion commune de l’équipe pour la biologie signifie que ses membres sont également intéressés par les résultats obtenus avec les algorithmes développés. En d’autres termes, si la principale expertise de l’équipe se situe au niveau méthodologique, BAOBAB est intéressée aussi par les questions biologiques sous-jacentes. Cet intérêt est poursuivi entre membres de l’équipe et en collaboration externe avec des chercheurs en biologie proches et lointains.
Outre les relations très étroites que BAOBAB maintient avec divers membres du projet HELIX et du LBBE externes à BAOBAB, l’équipe collabore ainsi, de façon informelle ou dans le contexte de projets financés aux niveaux national et international, avec des chercheurs dans de nombreux autres laboratoires de recherche en France et dans le monde. Les collaborations formelles se font également dans le cadre de co-encadrements de thèse. En particulier, tout un pan des activités de BAOBAB est poursuivi avec des étudiants ou ex-étudiants hors site. Il s’agit notamment de tout ce qui concerne la recherche et l’inférence de motifs dans les séquences biologiques et dans les structures (d’ARN et de protéines). Pour cette raison, ces travaux ne sont pas décrits dans ce rapport. Les personnes intéressées pourront trouver quelques informations à leur propos en visitant le site de BAOBAB. Ce réseau assez étendu de collaborations reflète le désir de l’équipe BAOBAB de maintenir une structure très ouverte sur le monde et sur les disciplines où elle ne possède pas d’expertise mais qui sont importantes à des degrés divers pour ses domaines actuels et futurs de recherche.
2) Structure et modèles d’évolution des séquences génomiques
(L. Canet, P. G. Fonseca, C. Gautier, L. Guéguen, C. Melo de Lima, V. Navratil , L. Palmeira-Gonon, M.F. Sagot, E. Tannier)
La compréhension de l’origine de la structuration d’un génome en régions homogènes (par exemple, isochores, tachychores) représente une des plus anciennes préocupations du laboratoire. Ce thème de recherche est abordé conjointement par l’équipe BGE (cf. IV) et par l’équipe BAOBAB. L’équipe BAOBAB s’est plus particulièrement intéressée au développement de méthodes permettant d’aborder cette question.
Dans le cadre d’une thèse en cours (Melo de Lima), un modèle de Markov caché a ainsi été mis en place pour prédire et analyser la structure en isochores des chromosomes humains. Une méthode de discrimination entre modèles de Markov a montré que certaines propriétés biologiques intervenaient pour classer un gène dans une classe d’isochores indépendamment du contenu en G+C de la séquence. Une étude complémentaire sur le génome du Tetraodon a confirmé ces résultats. Parmi les perspectives, nous envisageons de comparer, à partir de la méthode développée, l’organisation des isochores entre espèces afin d’essayer d’expliquer leur évolution au cours du temps. Ces travaux ont été conduits avec, en particulier, Laurent Guéguen, qui a rendu disponible via le web une bibliothèque de modules en langage Python (Sarment) regroupant des méthodes de partitionnement maximalement prédictif et de segmentation markovienne de séquences. Cette bibliothèque est décrite dans Guéguen, 2005.
Par ailleurs, une procédure de détection in silico de SNPs (« Single Nucleotide Polymorphisms ») à partir de données d’ESTs (« Expressed Sequence Tags ») a été mise au point dans le cadre de la thèse de Vincent Navratil (DigiPiNS ) (Digital SNP). À l’aide d’une base de connaissance réalisée et maintenue également par Vincent Navratil (voir la section « Dynamique des génomes »), il a ainsi été possible de procéder à la detection et à la caractérisation de plusieurs centaines de SNPs codants spécifiquement associés au phénotype tumoral chez l’homme (Aouacheria et al., 2005). L’objectif est maintenant 1. d’étudier de manière approfondie les facteurs influençant le spectre de distribution des fréquences alléliques pour chacun des génomes étudiés et 2. d’étudier la distribution et le niveau de conservation du polymorphisme entre des espèces proches.
Enfin, l´équipe s’intéresse également à la modélisation de la composition en mots de différents génomes, en lien avec des informations phylogénétiques, dans le but ultime d’estimer, par des méthodes d’inférence bayésienne, les influences de voisinage dans les processus de substitution (Thèse Palmiera). La thèse s’effectue en étroite collaboration avec l’équipe Bioinformatique et Génomique Évolutive (BGE) du LBBE puisqu’elle est co-encadrée par un membre de BAOBAB (Laurent Guéguen) et un membre de BGE (Jean Lobry).
3) Dynamique des génomes
(F. Boyer, M. D. V. Braga, C. Gautier, V. Lacroix, C. Lemaitre, V. Navratil, M.-F . Sagot, E. Tannier)
Au cours de l’évolution, l’organisation spatiale d’un génome subit divers types de remaniements dont la fréquence et les mécanismes sont encore relativement mal connus et compris. Nous nous intéressons à ces remaniements à la fois pour mieux en appréhender les processus biologiques dont ils sont la conséquence, et pour les utiliser afin d’étudier l’évolution structurelle et fonctionnelle des génomes d’organismes procaryotes et eucaryotes. Deux questions nous ont particulièrement intéressées jusqu’à présent : le calcul de distances de remaniements entre génomes et la détection de « segments conservés par des remaniements ».
La première question pose le problème du modèle d’évolution à adopter. Ce modèle doit prendre en compte la fréquence de chaque type de remaniement ainsi que d’autres paramètres (effet de la localisation génomique, etc.) que nous maîtrisons peu à l’heure actuelle. Parmi ceux-ci, un paramètre de fonction, intimement lié à la notion de segments conservés, devrait également être considéré. Notre travail jusqu’à maintenant a porté sur : 1. l’établissement et le maintien par Vincent Navratil d’une base de connaissances dédiée à la cartographie comparée des génomes de vertébrés (GemCore) initiée au cors d la thèse de G. Bronner ; 2. l’étude théorique d’un calcul de distance particulière (la distance d’inversion) entre génomes (Tannier et al., 2005 ; Tannier and Sagot, 2004) 3. l’étude théorique du lien entre distance d’inversion et segments conservés par remaniement (Sagot and Tannier, 2005) ; 4. une étude à la fois théorique et pratique sur des données eucaryotes dont le but à moyen et long terme est d’arriver à une meilleure compréhension des mécanismes moléculaires sous-jacents aux remaniements (thèse Lemaitre).
L’idée est d’arriver à avancer dans notre compréhension des mécanismes de remaniement, et des contraintes diverses agissant sur ces mécanismes, par un examen direct de la séquence génomique, en particulier des « régions de cassure », c’est-à-dire des régions situées autour des points de remaniement. L’objectif est de réussir à identifier, au niveau de la séquence, des caractéristiques propres à chaque type de remaniement et de parvenir ainsi à de meilleurs modèles de ces remaniements et des contraintes qui leur sont associées. Un panel de routines algorithmiques (CASSIS) est déjà disponible sous forme prototypale. Les analyses poursuivies font, entre autres, appel à l’expertise développée au sein de l’équipe sur les modèles de Markov et d’évolution de séquences (voir section « Structure et modèles d’évolution des séquences génomiques »).
4) Modélisation et analyse du métabolisme
(F. Boyer, P. G. Fonseca, V. Lacroix, M.-F . Sagot, E. Tannier, P. Thébault, L. Cottret)
Un des axes de l’équipe s’intéresse à la modélisation et à l’analyse du métabolisme des organismes procaryotes et eucaryotes (thèse Lacroix). Les objectifs principaux sont d’arriver à une meilleure compréhension de l’évolution du métabolisme et à relier cette évolution à celle de l’organisation spatiale ainsi qu’à la dynamique des génomes (voir section « Dynamique des génomes »).
La question à laquelle nous nous sommes plus particulièrement intéressés dans un premier temps concerne la recherche et l’inférence de modules et de motifs dans les réseaux métaboliques. Bien qu’aucune définition satisfaisante n’existe à l’heure actuelle, modules et motifs peuvent être vus comme les « briques de base » du métabolisme, briques évolutives ou fonctionnelles, les deux notions sont sans doute intimement liées. Selon le point de vue adopté, la différence entre ces deux notions peut d’ailleurs ne concerner que leur taille, les motifs étant considérés comme “petits” alors qu’une définition possible pour les modules les assimilerait aux voies métaboliques bien connues des biochimistes.
Figure 4 : exemple d’un motif défini comme un transformateur, c’est-à-dire comme un ensemble de fonctions enzymatiques. Ce motif apparaît répété quatre fois dans le réseau; l’une de ces occurrences est « à cheval » sur deux voies métaboliques.
Nous nous sommes penchés d’abord sur la question de la représentation du métabolisme et des réseaux métaboliques, adoptant pour l’heure un modèle statique sous forme de graphe (Figure 4). Ce modèle a l’inconvénient de négliger l’aspect quantitatif, et donc aussi dynamique de ces réseaux mais présente l’avantage d’en permettre une étude exploratoire exhaustive. Nous nous sommes également concentrés pour l’heure sur la brique la plus petite : les motifs (Lacroix et al., 2005). Un motif est identifié non à un élément purement topologique dans le graphe mais à un transformateur qui apparaîtrait répété de manière approximative, c’est-à-dire serait conservé au sein d’un même organisme, ou entre organismes différents. Un exemple est donné dans la Figure 4. Un prototype de logiciel (MOTUS), est également déjà disponible et se trouve en cours d’interfaçage pour une utilisation plus aisée par des biologistes (Cottret). Tous nos travaux concernant les réseaux métaboliques (Boyer post-doc) reposent fortement sur deux bases de connaissances, GEB et MicroBi, développées par nos collègues du Projet HELIX.
Outre le lien avec l’organisation et la dynamique génomiques (voir la section « Dynamique des génomes »), nos perspectives sur ce sujet concernent : 1. l’étude statistique des motifs en réseaux ; 2. l’introduction progressive de données quantitatives dans nos réseaux métaboliques ; 3. la prise en compte des données de régulation (cela va également dans le sens du point 2) ; 4. l’extension de notre étude à d’autres types de réseaux biochimiques (cela est une conséquence des points 1 et 2). Pour ce qui concerne le point 4, nous ferons appel, entre autres, à l’expertise qui se développe fortement au sein de l’équipe depuis environ un an autour des données d’expression et de leur analyse (co-encadrement par Christian Gautier des thèses de M. Tournoud et C. Truntzer, équipe Biostatistiques Santé).
5) Systèmes dynamiques
(E. Billoir, S. Charles, A. Chaumot, G. Decelière)
Tous les thèmes de recherche précédemment décrits ne prennent en compte, ou n’abordent la dynamique des systèmes biologiques que de façon indirecte, par une analyse, partielle, de l’effet et des conséquences de cette dynamique a posteriori. La modélisation directe de cette dynamique avait ainsi jusqu’à présent été laissée pour une deuxième étape, bien plus à long terme.
Ce long terme s’est très considérablement rapproché, début 2005, lorsque l’équipe a eu l’énorme chance de pouvoir accueillir en son sein Sandrine Charles, précédemment de l’équipe de Pierre Auger (au LBBE jusqu’en fin 2004). Cette chance est d’autant plus importante pour BAOBAB que Sandrine Charles apporte à l’équipe des connaissances et une expertise en systèmes dynamiques, écologie, dynamique des populations et modélisation de données spatiales en biologie. Cela permettra à BAOBAB de poursuivre son avance déjà amorcée vers l’étude méthodologique de systèmes de plus en plus complets et complexes.
De leur côté, les travaux de Sandrine allaient également dans le sens d’un rapprochement avec la génétique qui sera facilité grâce à sa récente intégration dans l’équipe BAOBAB. Ces travaux concernent des modèles de dynamique des populations développés initialement dans le cadre d’études en écotoxicologie.
Compte tenu de sa complexité, l’étude de la dynamique des populations en environnement hétérogène se heurte aujourd’hui au choix d’un formalisme mathématique adapté (systèmes dynamiques, modèles matriciels en temps discrets, équations aux dérivées partielles), et pose des questions d’ordre méthodologique comme : 1. l’intégration des différents niveaux d’organisation écologique (individus, populations, communautés, écosystèmes) dans les processus de modélisation et de simulation, ou/et 2. la prise en compte de la structuration spatiale des populations en assemblages de populations locales dont la dynamique est pour partie dépendante de processus de dispersion d’individus entre habitats favorables ou défavorables.
Plusieurs modèles matriciels de type Leslie, intégrant des modèles d’effet au niveau individuel, et permettant de prendre en compte une répartition géographique des individus entre habitats, ont ainsi été développés (Thèse Chaumot) : 1. pour étudier les effets de l’introduction d’un polluant dans une rivière sur la dynamique de populations de chironomes (Charles et al., 2004 ; Lopes et al., 2005) ; 2. pour modéliser la réponse à des perturbations environnementales d’une population de truites spatialement structurée (Chaumot et al., 2002 ; Chaumot et al., 2003a ; Chaumot et al., 2003b). De tels modèles ont également permis des applications en génétique des populations (Décelière et al., 2005) (thèse en collaboration avec C. Biémont). La thèse d’Émilie Vautrin sera l’occasion de poursuivre dans cette voie avec le développement de modèles pour des systèmes multi-infectés à transmission verticale (en collaboration avec F. Vavre).
Ce sont ces modèles également, seuls ou en association avec d’autres (de Markov, bayésiens) développés dans BAOBAB (voir, en particulier, la section « Structure et modèles d’évolution des séquences génomiques »), qui permettront à terme à BAOBAB d’introduire directement les aspects dynamiques, internes à un organisme et en lien avec son environnement (macro-évolution), dans les autres travaux en cours de l’équipe, notamment ceux décrits dans les sections « Dynamique des génomes » et « Modélisation et analyse du métabolisme ».
6- Perspectives
Les perspectives de BAOBAB incluent une poursuite des travaux menés, depuis plusieurs années ou depuis plus récemment, sur chacun des quatre thèmes décrits précédemment, avec souvent une expansion des sujets traités à l’intérieur de chacun de ces thèmes. Ainsi, par exemple, les travaux portant sur la modélisation et l’analyse du métabolisme devront être étendus prochainement à d’autres types de réseaux biologiques, notamment les réseaux génétiques et de signalisation, et à la prise en compte d’autres types de données, notamment dynamiques, concernant ces réseaux.
De fait, l’objectif à moyen et long terme est d’arriver, à travers une approche comparative à une meilleure compréhension de l’extraordinaire diversité des modes d’évolution et de fonctionnement des organismes (procaryotes et eucaryotes). Cette diversité semble reposer non seulement sur une diversité des éléments composant le vivant (gènes, métabolites, hormones, etc.) mais aussi, de manière non négligeable, sur l’extraordinaire combinatoire des interactions spatiales et temporelles possibles entre ces éléments, et de ces derniers, individuellement ou collectivement, avec leur environnement. Une compréhension du réseau complexe de ces interactions oblige à adopter un point de vue à la fois local et global, statique et dynamique, d’un organisme, allant du niveau moléculaire jusqu’à une vision générale du fonctionnement d’une cellule. Dans les prochaines années, nous nous intéresserons à chacun des niveaux séparément, depuis le génome jusqu’à la cellule, en mettant l’accent sur les liens entre ces différents niveaux.
Plus spécifiquement, ce sont essentiellement trois questions biologiques que nous souhaitons aborder : 1. existe-t-il des régularités, structurales ou fonctionnelles, dans la diversité qui est observée, régularités qui pourraient représenter des indices d’une organisation profonde du vivant ; 2. est-ce que nous pouvons identifier ces régularités de façon systématique et arriver ainsi à dégager un ordre dans le réseau complexe des interactions détectées ; enfin, 3. comment ce réseau s’est-il mis en place au cours de l’évolution, pour accomplir quelles fonctions, et aurait-il pu évoluer différemment ?
Cette étude sera abordée, comme elle l’a été dans le passé, par le développement de modèles et de méthodes d’analyse divers (statistiques, combinatoires, etc.). Ces modèles et méthodes seront systématiquement explorés et confrontés dans un esprit agnostique dans le but de répondre aux trois questions ci-dessus. Développements méthodologiques et meilleure compréhension de phénomènes biologiques seront ainsi deux processus qui avanceront côte à côte, ensemble. Les résultats attendus se situent d’abord par rapport à chacun de ces processus pris individuellement : 1. obtention de meilleurs formalismes de modélisation mathématique et algorithmes d’analyse ; et 2. réponse à la fois à des questions spécifiques (tests d’hypothèses) et plus générales (exploration systématique des données disponibles). La confrontation simultanée de nos tentatives de modélisation avec les données biologiques devrait également, surtout, permettre de mieux appréhender la structure des systèmes vivants : est-ce que cette structure est simple, ou simplifiable en des principes généraux, ou bien est-ce que la vie est faite essentiellement d’exceptions ?
Perspectives du département
Le département de Génétique et Génomique Évolutives vise à comprendre l’évolution et le fonctionnement des systèmes biologiques, en intégrant à la fois les connaissances sur les paramètres populationnels (sélection, dérive, migration) et sur les processus moléculaires (expression des gènes, mutations du matériel génétique) qui sont à la base de l’adaptation des espèces. Les projets des différentes équipes du département pour les années à venir font apparaitre plusieurs grandes tendances.
Tout d’abord, un effort important est mis sur l’identification des bases moléculaires de l’adaptation des espèces aux variations de leur environnement (biotique ou abiotique), au sein de systèmes hôtes-parasites complexes (incluant des insectes parasitoïdes, des bactéries endosymbiotiques et des virus). Cet axe implique des développements d’outils moléculaires nouveaux au laboratoire pour l’analyse du transcriptome et du protéome.
Par ailleurs, l’étude de l’évolution des génomes est révolutionnée grâce à la disponibilité d’un nombre toujours plus important de séquences de génomes complets de différentes espèces. En confrontant ces données de comparaisons interspécifiques avec des données de polymorphisme (disponibles publiquement, ou bien générées au laboratoire), il est désormais envisageable d’étudier en détail les processus d’évolution des séquences (chromosomes, gènes, éléments transposables, ...) à l’échelle de génomes entiers, et de déterminer la nature des forces évolutives (dérive, sélection, ...) à l’origine de cette évolution.
Enfin, il est clair que le catalogue des gènes d’un génome n’est pas sufffisant pour comprendre le fonctionnement d’un organisme. Une première étape dans la compréhension du lien entre génotype et phénotype consiste à étudier comment les gènes (ou leurs produits d’expression) interagissent entre eux. C’est pourquoi nous nous intéressons désormais à l’évolution des réseaux d’interaction biologiques.
Tous les projets développés au sein du département impliquent des développements méthodologiques importants. Trois axes méthodologiques majeurs peuvent êtres mentionnés :
- la modélisation mathématique de systèmes dynamiques (e.g. appliqués à la modélisation des interactions hôtes-parasites)
- le développement d’outils statistiques pour l’analyse de données (en particulier pour traiter les gros volumes de données génomiques, d’expression des gènes, ou pour l’analyse de la biodiversité)
- le développement d’algorithmes et de bases de données pour la génomique comparative (analyse de séquences, cartographie comparée, grands alignements génomiques multiples, banques de données d’alignements et d’arbres phylogénétiques)
Ces développements constituent une part essentielle de notre recherche et sont toujours menés avec le souci de mettre les outils à la disposition de l’ensemble de la communauté scientifique (via des services web ou des logiciels).
Publications du département (*: avant intégration au laboratoire)
2002
P1. Allemand R. (2002) La formation des Entomologistes. Les nouvelles de l’Entomologie française 14: 2-7.
P2. Allemand R., Brustel H., and Clary J. (2002) Une espèce de Cerambycidae nouvelle pour la faune de France, Aegomorphus francottei Sama (Coleoptera). Bull. mens. Soc. linn. Lyon 71: 105-114.
*P3. Aouacheria A., Neel B., Bouaziz Z., Dominique R., Walchshofer N., Paris J., Fillion H., and Gillet G. (2002) Carbazolequinone induction of caspase-dependent cell death in Src-overexpressing cells. Biochem Pharmacol. 64: 1605-1616.
*P4. Aouacheria A., Ory S., Schmitt J.R., Rigal D., Jurdic P., and Gillet G. (2002) p60(v-src) and serum control cell shape and apoptosis via distinct pathways in quail neuroretina cells. Oncogene 21: 1171-1186.
P5. Borie N., Maisonhaute C., Sarrazin S., Loevenbruck C., and Biémont C. (2002) Tissue-specificity of 412 retrotransposon expression in Drosophila simulans and D. melanogaster. Heredity 89: 247-252.
P6. Boulétreau-Merle J. and Fouillet P. (2002) How to overwinter and be a founder : egg–retention phenotypes and mating status in Drosophila melanogaster. Evol. Ecol. 16: 309-332.
P7. Bronner G., Spataro B., and Gautier C. (2002) Cartographie génomique comparée chez les mammifères. Médecine Science 18: 767-774.
P8. Bronner G., Spataro B., Page M., Gautier C., and Rechenmann F. (2002) Modeling comparative mapping using objects and associations. Comput. Chem. 26: 413-420.
P9. Chaumot A., Charles S., Flammarion P., Garric J., and Auger P. (2002) Using aggregation methods to assess toxicant effects on population dynamics in spatial systems. Ecological Applications 12: 1771-1784.
P10. Chureau C., Prissette M., Bourdet A., Barbe V., Cattolico L., Jones L., Eggen A., Avner P., and Duret L. (2002) Comparative sequence analysis of the X-inactivation center region in mouse, human, and bovine. Genome Res. 12: 894-908.
P11. Culhane A.C., Perrière G., Considine E.C., Cotter T.G., and Higgins D.G. (2002) Between-group analysis of microarray data. Bioinformatics 18: 1600-1608.
P12. Daubin V., Gouy M., and Perrière G. (2002) A phylogenomic approach to bacterial phylogeny : evidence of a core of genes sharing a common history. Genome Res. 12: 1080-1090.
P13. Debout G., Fauvergue X., and Fleury F. (2002) The effect of foundress number on sex ratio under partial local mate competition. Ecological Entomology 27: 242-246.
P14. Duret L. (2002) Evolution of synonymous codon usage in metazoans. Curr. Opin. Genet. Dev. 12: 640-649.
P15. Duret L., Semon M., Piganeau G., Mouchiroud D., and Galtier N. (2002) Vanishing GC-Rich Isochores in Mammalian Genomes. Genetics 162: 1837-1147.
P16. Founoune H., Duponnois R., Meyer J.M., Thioulouse J., Masse D., Chotte J.L., and Neyra M. (2002) Interactions between ectomycorrhizal symbiosis and fluorescent pseudomonads on Acacia holosericea : isolation of mycorrhiza helper bacteria (MHB) from a Soudano-Sahelian soil. FEMS Microbiology Ecology 41: 37-46.
P17. Jarraud S., Mougel C., Thioulouse J., Lina G., Meugnier H., Forey F., Nesme X., Etienne J., and Vandenesch F. (2002) Relationships between Staphylococcus aureus genetic background, virulence factors, agr groups (alleles), and human disease. Infect. Immun. 70: 631-641.
*P18. Lalle P., Aouacheria A., Dumont-Miscopein A., Jambon M., Venet S., Bobichon H., Colas P., Deleage G., Geourjon C., and Gillet G. (2002) Evidence for crucial electrostatic interactions between Bcl-2 homology domains BH3 and BH4 in the anti-apoptotic Nr-13 protein. Biochem. J. 368: 213-221.
P19. Lepetit D., Brehm A., Fouillet P., and Biémont C. (2002a) Insertion polymorphism of retrotransposable elements in populations of the insular, endemic species Drosophila madeirensis. Mol. Ecol. 11: 347-354.
P20. Lerat E., Capy P., and Biémont C. (2002a) The relative abundance of dinucleotides in transposable elements in five species. Mol. Biol. Evol. 19: 964-967.
P21. Lerat E., Capy P., and Biémont C. (2002b) Codon usage by transposable elements and their host genes in five species. J. Mol. Evol. 54: 625-637.
P22. Lobry J.R. and Sueoka N. (2002) Asymmetric directional mutation pressures in bacteria. Genome Biol. 3: RESEARCH0058.
P23. Marais G. and Piganeau G. (2002) Hill-Robertson interference is a minor determinant of variations in codon bias across Drosophila melanogaster and Caenorhabditis elegans genomes. Mol. Biol. Evol. 19: 1399-1406.
P24. Mathé C., Sagot M.-F., Schiex T., and Rouzé P. (2002) Current methods of gene prediction, their strengths and weaknesses. Nucleic Acids Research 30: 4103-4117.
P25. Mathé C., Schiex T., Rouzé P., Blayo P., and Sagot M.-F. (2002) Gene finding in eukaryotes. In Gene cloning and expression technologies (eds. Lu Q. and Weiner M.), pp. 27-43. Eaton Publishing.
P26. Mougel C., Thioulouse J., Perrière G., and Nesme X. (2002) A mathematical method for determining genome divergence and species delineation using AFLP. International Journal of Systematic and Evolutionary Microbiology 57: 573-586.
P27. Perrière G. and Thioulouse J. (2002) Use and misuse of correspondence analysis in codon usage studies. Nucleic Acids Research 30: 4548-4555.
P28. Pétavy G., Moreteau B., Gibert P., and David J.R. (2002) Phenotypic plasticity of body pigmentation in Drosophila : influence of a developmental thermoperiodic regime in two sibling species. Physiological Entomology 27: 124-135.
P29. Piganeau G., Mouchiroud D., Duret L., and Gautier C. (2002) Expected Relationship Between the Silent Substitution Rate and the GC Content : Implications for the Evolution of Isochores. J. Mol. Evol. 54: 129-133.
P30. Pisanti N. and Sagot M.-F. (2002) Further thoughts on the syntenic distance between genomes. Algorithmica 34: 157-180.
P31. Ponger L. and Mouchiroud D. (2002) CpGProD : identifying CpG islands associated with transcription start sites in large genomic mammalian sequences. Bioinformatics 18: 631-633.
P32. Pontier D., Say L., Debias F., Bried J., Thioulouse J., Micol T., and Natoli E. (2002) The diet of feral cats (Felis catus L.) at five sites on the Grande Terre, Kerguelen archipelago. Polar Biology 25: 833-837.
P33. Rafalimanana H., Kaiser L., and Delpuech J.M. (2002) Stimulating effects of the insecticide chlorpyrifos on host searching and infestation efficacy of a parasitoid wasp. Pest Manag. Sci. 58: 321-328.
P34. Rizzon C., Marais G., Gouy M., and Biémont C. (2002) Recombination rate and the distribution of transposable elements in the Drosophila melanogaster genome. Genome Res. 12: 400-407.
P35. Tahi F., Gouy M., and Regnier M. (2002) Automatic RNA secondary structure prediction with a comparative approach. Comput. Chem 26: 521-530.
P36. Tavares R., Vidal J., van Lammeren A., and Kreis M. (2002) AtSKtheta, a plant homologue of SGG/GSK-3 marks developing tissues in Arabidopsis thaliana. Plant Mol. Biol. 50: 261-271.
P37. Tavares R., Vidal J., van Lammeren A., and Kreis M. (2002) Non-purified anti-peptide sera generate tissue specific artefacts in immunohistochemical staining of Arabidopsis thaliana. Plant Science 162: 309-314.
P38. Vavre F., Fleury F., Varaldi J., Fouillet P., and Boulétreau M. (2002) Infection polymorphism and cytoplasmic incompatibility in Hymenoptera-Wolbachia associations. Heredity 88: 361-365.
P39. Vieira C., Nardon C., Arpin C., Lepetit D., and Biémont C. (2002) Evolution of genome size in Drosophila. is the invader’s genome being invaded by transposable elements ? Mol. Biol. Evol. 19: 1154-1161.
P40. Vivares C.P., Gouy M., Thomarat F., and Metenier G. (2002) Functional and evolutionary analysis of a eukaryotic parasitic genome. Curr. Opin Microbiol. 5: 499-505.
P41. Zaoui S. and Biémont C. (2002) [Frequency of consanguineous unions in the Tlemcen area (West Algeria)]. Santé 12: 289-295.
2003
P42. Allemand R. (2003) Checklist of the French Oedemeridae-Liste des Oedemeridae de la faune de France (Coleoptera).). Bull. mens. Soc. linn. Lyon, 72: 229-232.
P43. Allemand R., Lemaître C., Frey F., Boulétreau M., Vavre F., Nordlander G., van Alphen J., and Carton Y. (2003) Phylogeny of six African Leptopilina species (Hymenoptera : Cynipoidea, Figitidae), parasitoids of Drosophila spp., with descriptions of three new species. Annales de la Société Entomologique de France 38: 319-332.
*P44. Aouacheria A., Banyai M., Rigal D., Schmidt C.J., and Gillet G. (2003) Characterization of vnr-13, the first alphaherpesvirus gene of the bcl-2 family. Virology 316: 256-266.
P45. Assossou O., Besson F., Rouault J.P., Persat F., Brisson C., Duret L., Ferrandiz J., Mayencon M., Peyron F., and Picot S. (2003) Subcellular localization of 14-3-3 proteins in Toxoplasma gondii tachyzoites and evidence for a lipid raft-associated form. Fems. Microbiol. Lett. 224: 161-118.
P46. Benit L., Calteau A., and Heidmann T. (2003) Characterization of the low-copy HERV-Fc family : evidence for recent integrations in primates of elements with coding envelope genes. Virology 312: 159-168.
P47. Bieche I., Laurent A., Laurendeau I., Duret L., Giovangrandi Y., Frendo J.L., Olivi M., Fausser J.L., Evain-Brion D., and Vidaud M. (2003) Placenta-Specific INSL4 Expression Is Mediated by a Human Endogenous Retrovirus Element. Biol. Reprod. 68: 1422-1149.
P48. Biémont C., Nardon C., Déceliere G., Lepetit D., Loevenbruck C., and Vieira C. (2003) Worldwide distribution of transposable element copy number in natural populations of Drosophila simulans. Evolution Int. J. Org. Evolution 57: 159-167.
P49. Blayo P., Rouzé P., and Sagot M.-F. (2003) Orphan gene finding : An exon assembly approach. Theoretical Computer Science 290: 1407-1431.
P50. Boulétreau-Merle J., Fouillet P., and Varaldi J. (2003) Divergent strategies in low temperature environment for the sibling species Drosophila melanogaster and D. simulans : Overwintering in extension border areas of France and comparison with African populations. Evolutionary Ecology 17: 523-548.
P51. Cadet P., Pate E., and Thioulouse J. (2003) Relationship of nematode communities to human demographics and environment in agricultural fields and fallow lands in Senegal. Journal of Tropical Ecology 19: 279-290.
P52. Charles-Bajard S., Flandrois J.-P., and Tomassone R. (2003) Quelques problèmes statistiques rencontrés dans l'estimation de la température minimale de croissance de Listeria monocytogenes. Revue de Statistique Appliquée L1, 59-71.
P53. Charles S., Ney M., Mouchiroud D., Humblot L., and Batier C. (2003) Un site web pour l'enseignement interdisciplinaire des Mathématiques en Biologie. In EIAH 2003 (Environnement Informatique d'Apprentissage Humain), pp. 445-452.
P54. Chaumot A., Charles S., Flammarion P., and Auger P. (2003a) Do migratory or demographic disruptions rule the population impact of pollution in spatial networks? Theoretical Population Biology 64: 473-480.
P55. Chaumot A., Charles S., Flammarion P., and Auger P. (2003b) Ecotoxicology and spatial modeling in population dynamics: an illustration with brown trout. Environmental Toxicology and Chemistry 22: 958-969.
P56. Culhane A.C., Perrière G., and Higgins D.G. (2003) Cross-platform comparison and visualisation of gene expression data using co-inertia analysis. BMC Bioinformatics 4: 59.
P57. Daubin V., Lerat E., and Perrière G. (2003) The source of laterally transferred genes in bacterial genomes. Genome Biol. 4: R57.
*P58. Daubin V., Moran N.A., and Ochman H. (2003) Phylogenetics and the cohesion of bacterial genomes. Science 301: 829-832.
P59. Daubin V. and Perrière G. (2003) G+C3 structuring along the genome : a common feature in prokaryotes. Mol. Biol. Evol. 20: 471-483.
P60. David J.R., Gibert P., Mignon-Grasteau S., Legout H., Petavy G., Beaumont C., and Moreteau B. (2003) Genetic variability of sexual size dimorphism in a natural population of Drosophila melanogaster : an isofemale-line approach. J. Genet. 82: 79-88.
P61. David J.R., Gibert P., Moreteau B., Gilchrist G., and Huey R.B. (2003) The fly that came in from the cold : geographic variation of recovery time from low-temperature exposure in Drosophila subobscura. Functional Ecology 17: 425-430.
P62. Dedeine F., Bandi C., Boulétreau M., and Kramer L.H. (2003) Insights into Wolbachia obligatory symbiosis. In Insect Symbiosis (ed. Miller K.B.T.), pp. 267-282. CRC Press LLC.
P63. Delpuech J.M. and Meyet J. (2003) Reduction in the sex ratio of the progeny of a parasitoid wasp (Trichogramma brassicae) surviving the insecticide chlorpyrifos. Arch. Environ. Contam. Toxicol. 45: 203-208.
P64. Devulder G., Perrière G., Baty F., and Flandrois J.P. (2003) BIBI, a bioinformatics bacterial identification tool. J. Clin. Microbiol. 41: 1785-1787.
P65. Dray S., Chessel D., and Thioulouse J. (2003a) Procustean co-inertia analysis for the linking of multivariate datasets. Ecoscience 10: 110-119.
P66. Dray S., Chessel D., and Thioulouse J. (2003b) Co-inertia analysis and the linking of ecological tables. Ecology 84: 3078-3089.
P67. Fabbri R. and Allemand R. (2003) Revision of the species of Pedilophorus of the group auratus (Duftschmid, 1825) and additionnal notes on the group macedonicus. Riv. Mus. Sc nat. Bergamo, 21: 3-29.
P68. Freeman J.C., Allemand R., and Van Meer C. (2003) Odontosphindus grandis Hampe, nouvelle espèce, nouveau genre, nouvelle sous-famille pour la faune de France et pour l’Europe occidentale (Coleoptera Sphindidae). Bull. Soc. Ent. Fr. 108: 221-232.
P69. Grassot J., Mouchiroud G., and Perrière G. (2003) RTKdb : database of Receptor Tyrosine Kinase. Nucleic Acids Res. 31: 353-358.
P70. Haerty W., Gibert P., Capy P., Moreteau B., and David J.R. (2003) Microspatial structure of Drosophila melanogaster populations in Brazzaville : evidence of natural selection acting on morphometrical traits. Heredity 91: 440-447.
*P71. Lerat E., Daubin V., and Moran N.A. (2003) From gene trees to organismal phylogeny in prokaryotes: the case of the gamma-Proteobacteria. PLoS Biol. 1: E19.
P72. Lerat E., Rizzon C., and Biémont C. (2003) Sequence divergence within transposable element families in the Drosophila melanogaster genome. Genome Res. 13: 1889-1896.
P73. Lobry J.R. and Chessel D. (2003) Internal correspondence analysis of codon and amino-acid usage in thermophilic bacteria. J. Appl. Genet. 44: 235-261.
P74. Lobry J.R. and Louarn J.M. (2003) Polarisation of prokaryotic chromosomes. Curr. Opin. Microbiol. 6: 101-108.
*P75. Marais G. (2003) Biased gene conversion : implications for genome and sex evolution. Trends Genet. 19: 330-338.
*P76. Marais G. and Charlesworth B. (2003) Genome evolution: recombination speeds up adaptive evolution. Curr. Biol. 13: R68-70.
*P77. Marais G. and Galtier N. (2003) Sex chromosomes : how X-Y recombination stops. Curr. Biol. 13: R641-643.
P78. Marais G., Mouchiroud D., and Duret L. (2003) Neutral effect of recombination on base composition in Drosophila. Genet. Res. 81: 79-89.
P79. Moreteau B., Gibert P., Delpuech J.M., Petavy G., and David J.R. (2003) Phenotypic plasticity of sternopleural bristle number in temperate and tropical populations of Drosophila melanogaster. Genet. Res. 81: 25-32.
P80. Moreteau B., Gibert P., Pétavy G., Moreteau J.C., Huey R.B., and David J.R. (2003) Morphometrical evolution in a Drosophila clade : the Drosophila obscura group. Journal of Zoological Systematics and Evolutionary Research 41: 64-71.
P81. Mouton L., Henri H., Boulétreau M., and Vavre F. (2003) Strain-specific regulation of intracellular Wolbachia density in multiply infected insects. Mol. Ecol. 12: 3459-3465.
P82. Nardon C., Weiss M., Vieira C., and Biémont C. (2003) Variation of the genome size estimate with environmental conditions in Drosophila melanogaster. Cytometry A 55: 43-49.
P83. Ollier S., Chessel D., Couteron P., Pélissier R., and Thioulouse J. (2003) Comparing and classifying one-dimensional spatial patterns : an application to laser altimeter profiles. Remote Sensing of Environment 85: 453-462.
P84. Perrière G., Combet C., Penel S., Blanchet C., Thioulouse J., Geourjon C., Grassot J., Charavay C., Gouy M., Duret L., and Deléage G. (2003) Integrated databanks access and sequence/structure analysis services at the PBIL. Nucleic Acids Research 31: 3393-3399.
P85. Perrière G. and Thioulouse J. (2003) Use of Correspondence Discriminant Analysis to predict the subcellular location of bacterial proteins. Computer Methods and Programs in Biomedicine 70: 99-105.
P86. Philippe H., Casane D., Gribaldo S., Lopez P., and Meunier J. (2003) Heterotachy and functional shift in protein evolution. IUBMB Life 55: 257-265.
P87. Pisanti N., Crochemore M., Grossi R., and Sagot M.-F. (2003) A basis of tiling motifs for generating repeated patterns and its complexity for higher quorum. In 28th International Symposium on Mathematical Foundations of Computer Science, pp. 622-632. Springer Verlag.
P88. Rizzon C., Martin E., Marais G., Duret L., Segalat L., and Biémont C. (2003) Patterns of selection against transposons inferred from the distribution of Tc1, Tc3, and Tc5 insertions in the mut-7 line of the nematode Caenorhabditis elegans. Genetics 165: 1127-1135.
P89. Robin S., Daudin J.-J., Richard H., Sagot M.-F., and Schbath S. (2003) Occurrence probability of structured motifs in random sequences. J. Computational Biology 9: 761-773.
P90. Sagot M.-F. and Wakabayashi Y. (2003) Pattern inference under many guises. In Recent advances in algorithms and combinatorics (eds. Reed B. and Sales C.L.), pp. 245-288. Springer Verlag.
P91. Varaldi J., Fouillet P., Ravallec M., Lopez-Ferber M., Boulétreau M., and Fleury F. (2003) Infectious behavior in a parasitoid. Science 302: 1930.
P92. Vavre F., Fouillet P., and Fleury F. (2003) Between- and within-host species selection on cytoplasmic incompatibility-inducing Wolbachia in haplodiploids. Evolution Int. J. Org. Evolution 57: 421-427.
P93. Vogel J., Normand P., Thioulouse J., Nesme X., and Grundmann G. (2003) Relationship between spatial and genetic distance in Agrobacterium spp. in 1 cubic centimeter of soil. Applied and Environmental Microbiology 69: 1482-1487.
2004
P94. Allali J. and Sagot M.-F. (2004) Novel tree edit operations for RNA secondary structure comparison. In WABI’04, pp. 412-425. Springer Verlag.
P95. Allemand R. (2004) Révision des Simplocaria Stephens, 1830, de la région sud-ouest paléarctique (Coleoptera Byrrhidae). Bull. Soc. Ent. Fr 109: 375-377.
P96. Aouacheria A., Cluzel C., Lethias C., Gouy M., Garrone R., and Exposito J.Y. (2004) Invertebrate data predict an early emergence of vertebrate fibrillar collagen clades and an anti-incest model. J. Biol. Chem. 279: 47711-47719.
P97. Ayrinhac A., Debat V., Gibert P., Kister A.-G., Legout H., Moreteau B., Vergilino R., and David J. (2004) Cold adaptation in geographical populations of Drosophila melanogaster: phenotypic plasticity is more important than genetic variability. Functional Ecology 18: 700-706.
P98. Baines J.F., Das A., Mousset S., and Stephan W. (2004) The role of natural selection in genetic differentiation of worldwide populations of Drosophila ananassae. Genetics 168: 1987-1998.
P99. Belle E., Duret L., Galtier N., and Eyre-Walker A. (2004) The Decline of Isochores in Mammals : An Assessment of the GC Content Variation Along the Mammalian Phylogeny. J. Mol. Evol. 58: 653-660.
P100. Biémont C., Monti-Dedieu L., and Lemeunier F. (2004) Detection of transposable elements in Drosophila salivary gland polytene chromosomes by in situ hybridization. Methods Mol. Biol. 260: 21-28.
P101. Biémont C. and Vieira C. (2004) L’influence des éléments transposables sur la taille des génomes. J. Soc. Biol. 198: 413-417.
P102. Bonnaud B., Bouton O., Oriol G., Cheynet V., Duret L., and Mallet F. (2004) Evidence of Selection on the Domesticated ERVWE1 env Retroviral Element Involved in Placentation. Mol. Biol. Evol. 21: 1895-1901.
P103. Bonnefoy-Berard N., Aouacheria A., Verschelde C., Quemeneur L., Marcais A., and Marvel J. (2004) Control of proliferation by Bcl-2 family members. Biochim. Biophys. Acta 1644: 159-168.
P104. Boussau B., Karlberg E.O., Frank A.C., Legault B.A., and Andersson S.G. (2004) Computational inference of scenarios for alpha-proteobacterial genome evolution. Proc. Natl. Acad. Sci. U.S.A. 101: 9722-9727.
P105. Capy P. and Gibert P. (2004) Drosophila melanogaster, Drosophila simulans : so similar yet so different. Genetica 120: 5-16.
P106. Carvalho A., Freitas A.T., Oliveira A.L., and Sagot M.-F. (2004) A parallel algorithm for the extraction of structured motifs. In ACM Symposium on Applied Computing (SAC’04), pp. 147-153. ACM Press.
P107. Carvalho A., Freitas A.T., Oliveira A.L., and Sagot M.-F. (2004) Efficient Extraction of Structured Motifs Using Box-links. In SPIRE’04, pp. 267-268. Springer Verlag.
P108. Charles S., Ferreol M., Chaumot A., and Péry A.R.R. (2004) Food availability effect on population dynamics of the midge Chironomus riparius : a Leslie modeling approach. Ecological Modelling 175: 217-229.
P109. Chessel D., Dufour A.B., and Thioulouse J. (2004) The ade4 package - I : One-table methods. Rnews 4: 5-10.
P110. Crochemore M., Giancarlo R., and Sagot M.-F. (2004) Longest motifs with a functionally equivalent block. In SPIRE’04, pp. 298-209. Springer Verlag.
P111. Crochemore M., Iliopoulos C., Mohamed M., and Sagot M.-F. (2004) Longest Repeated Motif with k Don’t Cares. In LATIN’04, pp. 271-278. Springer Verlag.
*P112. Daubin V. and Moran N.A. (2004) Comment on « The origins of genome complexity ». Science 306: 978; author reply 978.
*P113. Daubin V. and Ochman H. (2004a) Start-up entities in the origin of new genes. Curr. Opin. Genet. Dev 14: 616-619.
*P114. Daubin V. and Ochman H. (2004b) Bacterial genomes as new gene homes: the genealogy of ORFans in E. coli. Genome Res. 14: 1036-1042.
*P115. Daubin V. and Ochman H. (2004) Quartet mapping and the extent of lateral transfer in bacterial genomes. Mol. Biol. Evol. 21: 86-89.
P116. David J.R., Allemand R., Capy P., Chakir M., Gibert P., Petavy G., and Moreteau B. (2004) Comparative life histories and ecophysiology of Drosophila melanogaster and D. simulans. Genetica 120: 151-163.
P117. Dedeine F., Vavre F., Shoemaker D.D., and Boulétreau M. (2004) Intra-individual coexistence of a Wolbachia strain required for host oogenesis with two strains inducing cytoplasmic incompatibility in the wasp Asobara tabida. Evolution Int. J. Org. Evolution 58: 2167-2174.
P118. Delaunay L. and Allemand R. (2004) Hypera denominonda Capiomont, espèce nouvelle pour la faune de France (Coleoptera Curculionidae). Bull. mens. Soc. linn. Lyon 73: 209-212.
P119. Fleury F., Ris N., Allemand R., Fouillet P., Carton Y., and Boulétreau M. (2004) Ecological and genetic interactions in Drosophila-parasitoids communities : a case study with D. melanogaster, D. simulans and their common Leptopilina parasitoids in south-eastern France. Genetica 120: 181-194.
P120. Gautier C., Navratil V., and Melo de Lima C. (2004) Genomic Information along the Genome: Modeling Statistical analysis and Evolutionary Implication IFIP World Computer Congress (18th) 22-27 Aug. 2004
P121. Gavotte L., Vavre F., Henri H., Ravallec M., Stouthamer R., and Boulétreau M. (2004) Diversity, distribution and specificity of WO phage infection in Wolbachia of four insect species. Insect Mol. Biol. 13: 147-153.
P122. Gibert P., Capy P., Imasheva A., Moreteau B., Morin J.P., Petavy G., and David J.R. (2004b) Comparative analysis of morphological traits among Drosophila melanogaster and D. simulans: genetic variability, clines and phenotypic plasticity. Genetica 120: 165-179.
P123. Gibert P., Moreteau B., and David J.R. (2004a) Phenotypic plasticity of body pigmentation in Drosophila melanogaster : genetic repeatability of quantitative parameters in two successive generations. Heredity 92: 499-507.
P124. Ginolhac A., Jarrin C., Gillet B., Robe P., Pujic P., Tuphile K., Bertrand H., Vogel T.M., Perrière G., Simonet P., and Nalin R. (2004) Phylogenetic analysis of polyketide synthase I domains from soil metagenomic libraries allows selection of promising clones. Appl. Environ. Microbiol. 70: 5522-5527.
P125. Jacq N., Blanchet C., Combet C., Cornillot E., Duret L., Kurata K., Nakamura H., Silvestre T., and Breton V. (2004) Grid as a bioinformatic tool. Parallel Computing 30: 1093-1107.
P126. Jaillon O., Aury J.M., Brunet F., Petit J.L., Stange-Thomann N., Mauceli E., Bouneau L., Fischer C., Ozouf-Costaz C., Bernot A., Nicaud S., Jaffe D., Fisher S., Lutfalla G., Dossat C., Segurens B., Dasilva C., Salanoubat M., Levy M., Boudet N., Castellano S., Anthouard V., Jubin C., Castelli V., Katinka M., Vacherie B., Biémont C., Skalli Z., Cattolico L., Poulain J., De Berardinis V., Cruaud C., Duprat S., Brottier P., Coutanceau J.P., Gouzy J., Parra G., Lardier G., Chapple C., McKernan K.J., McEwan P., Bosak S., Kellis M., Volff J.N., Guigo R., Zody M.C., Mesirov J., Lindblad-Toh K., Birren B., Nusbaum C., Kahn D., Robinson-Rechavi M., Laudet V., Schachter V., Quetier F., Saurin W., Scarpelli C., Wincker P., Lander E.S., Weissenbach J., and Roest Crollius H. (2004) Genome duplication in the teleost fish Tetraodon nigroviridis reveals the early vertebrate proto-karyotype. Nature 431: 946-957.
P127. Keime C., Damiola F., Mouchiroud D., Duret L., and Gandrillon O. (2004) Identitag, a relational database for SAGE tag identification and interspecies comparison of SAGE libraries. BMC Bioinformatics 5: 143.
P128. Le Roux F., Gay M., Lambert C., Nicolas J.L., Gouy M., and Berthe F. (2004) Phylogenetic study and identification of Vibrio splendidus-related strains based on gyrB gene sequences. Dis. Aquat. Organ. 58: 143-150.
P129. Lobry J.R. (2004) Life history traits and genome structure : aerobiosis and G+C content in bacteria. Lecture Notes in Computer Sciences 3039: 679-686.
P130. Mallet F., Bouton O., Prudhomme S., Cheynet V., Oriol G., Bonnaud B., Lucotte G., Duret L., and Mandrand B. (2004) The endogenous retroviral locus ERVWE1 is a bona fide gene involved in hominoid placental physiology. Proc. Natl. Acad. Sci. USA 101: 1731-1736.
*P131. Marais G., Charlesworth B., and Wright S.I. (2004) Recombination and base composition : the case of the highly self-fertilizing plant Arabidopsis thaliana. Genome Biol. 5: R45.
P132. Marais G., Domazet-Loso T., Tautz D., and Charlesworth B. (2004) Correlated evolution of synonymous and nonsynonymous sites in Drosophila. J. Mol. Evol. 59: 771-779.
P133. Meunier J. and Duret L. (2004) Recombination drives the evolution of GC-content in the human genome. Mol. Biol. Evol. 21: 984-990.
P134. Michon L., Morle L., Bozon M., Duret L., Zech J.C., Godet J., Plauchu H., and Edery P. (2004) Physical and transcript map of the autosomal dominant colobomatous microphthalmia locus on chromosome 15q12-q15 and refinement to a 4.4 Mb region. Eur. J. Hum Genet. 12: 574-578.
P135. Mignon-Grasteau S., David J., Gibert P., Legout H., Petavy G., Moreteau B., and Beaumont C. (2004) REML estimates of genetic parameters of sexual dimorphism for wing and thorax length in Drosophila melanogaster. J. Genet. 83: 163-170.
P136. Mousset S. and Derome N. (2004) Molecular polymorphism in Drosophila melanogaster and D. simulans : what have we learned from recent studies? Genetica 120: 79-86.
*P137. Mousset S., Derome N., and Veuille M. (2004) A test of neutrality and constant population size based on the mismatch distribution. Mol. Biol. Evol. 21: 724-731.
P138. Mouton L., Dedeine F., Henri H., Boulétreau M., Profizi N., and Vavre F. (2004) Virulence, multiple infections and regulation of symbiotic population in the Wolbachia-Asobara tabida symbiosis. Genetics 168: 181-189.
P139. Naizot T., Auda Y., Dervieux A., Thioulouse J., and Bellan M.F. (2004) Une nouvelle analyse multi-temporelle d’images satellitales, les residus de l’Analyse en Composantes Principales. Un cas d’etude: une serie d’images Landsat Thematic Mapper de la Camargue, France. Int. J. Remote Sensing 25: 1925-1938.
P140. Reymond N., Charles H., Duret L., Calevro F., Beslon G., and Fayard J.-M. (2004) ROSO: Research on Optimized Oligonucleotide Probes for Microarrays. Bioinformatics 20: 271-273.
P141. Ris N., Allemand R., Fouillet P., and Fleury F. (2004) The joint effect of temperature and host species induce complex genotype-by-environment interactions in the larval parasitoid of Drosophila, Leptopilina heterotoma (Hymenoptera Figitidae). Oikos 106: 451-456.
P142. Robinson-Rechavi M., Boussau B., and Laudet V. (2004) Phylogenetic dating and characterization of gene duplications in vertebrates : the cartilaginous fish reference. Mol. Biol. Evol. 21: 580-586.
P143. Ruau D., Duarte J., Ourjdal T., Perrière G., Laudet V., and Robinson-Rechavi M. (2004) Update of NUREBASE : nuclear hormone receptor functional genomics. Nucleic Acids Res. 32: D165-167.
P144. Sebo A. and Tannier E. (2004) On Metric Generators of Graphs. Mathematics of Operations Research 29: 383-393.
P145. Semon M. and Duret L. (2004) Evidence that functional transcription units cover at least half of the human genome. Trends Genet. 20: 229-232.
P146. Tannier E. and Sagot M.-F. (2004) Sorting by reversals in subquadratic time. In CPM’04, pp. 1-13. Springer Verlag.
P147. Thioulouse J., Simier M., and Chessel D. (2004) Simultaneous analysis of a sequence of paired ecological tables. Ecology 85: 272-283.
P148. Thomarat F., Vivares C.P., and Gouy M. (2004) Phylogenetic analysis of the complete genome sequence of Encephalitozoon cuniculi supports the fungal origin of microsporidia and reveals a high frequency of fast-evolving genes. J. Mol. Evol. 59: 780-791.
P149. Vavre F., de Jong J.H., and Stouthamer R. (2004) Cytogenetic mechanism and genetic consequences of thelytoky in the wasp Trichogramma cacoeciae. Heredity 93: 592-596.
P150. Vieira C. and Biémont C. (2004) Transposable element dynamics in two sibling species : Drosophila melanogaster and Drosophila simulans. Genetica 120: 115-123.
P151. Wuyts J., Perrière G., and Van De Peer Y. (2004) The European ribosomal RNA database. Nucleic Acids Res. 32: D101-103.
P152. Zelwer M. and Daubin V. (2004) Detecting phylogenetic incongruence using BIONJ : an improvement of the ILD test. Mol. Phylogenet. Evol. 33: 687-693.
2005
P153. Allali J. and Sagot M.-F. (2005) A new distance for high level RNA secondary structure comparison. IEEE/ACM Transactions on Computational Biology and Bioinformatics 2: 3-14.
P154. Allali J. and Sagot M.-F. (2005) A Multiple Graph Layers Model with Application to RNA Secondary Structures Comparison. In SPIRE’05.
P155. Allemand R. (2005) Ptilophorus (=Evaniocera) dufouri (Rhipiphoridae), un Coléoptère parasite encore bien secret ! Le Coléoptériste 7: 195-198.
P156. Aouacheria A, Brunet F, Gouy M. (2005) Phylogenomics of Life-or-Death Switches in Multicellular Animals: Bcl-2, BH3-only and BNip Families of Apoptotic Regulators.Mol Biol Evol.22 (12): 2395-2416
P157. Aouacheria A., Navratil V., Wen W., Jiang M., Mouchiroud D., Gautier C., Gouy M., and Zhang M. (2005) In silico whole-genome scanning of cancer-associated nonsynonymous SNPs and molecular characterization of a dynein light chain tumour variant. Oncogene 24: 6133-6142.
P158. Assigbetse K., Gueye M., Thioulouse J., and Duponnois R. (2005) Soil bacterial diversity responses to root colonization by an ectomycorrhizal fungus are not root-growth dependent. Microbial Ecology in press.
P159. Aurell H., Farge P., Meugnier H., Gouy M., Forey F., Lina G., Vandenesch F., Etienne J., and Jarraud S. (2005) Clinical and environmental isolates of Legionella pneumophila serogroup 1 cannot be distinguished by sequence analysis of two surface protein genes and three housekeeping genes. Appl Environ. Microbiol. 71: 282-289.
P160. Bazin E., Duret L., Penel S., and Galtier N. (2005) Polymorphix : a sequence polymorphism database. Nucleic Acids Res. 33 Database Issue: 481-484.
P161. Biémont C. and Vieira C. (2005) What transposable elements tell us about genome organization and evolution: the case of Drosophila. Cytogenet. Genome Res. 110: 25-34.
P162. Bonnaud B., Beliaeff J., Bouton O., Oriol G., Duret L., and Mallet F. (2005) Natural history of the ERVWE1 endogenous retroviral locus. Retrovirology 2: 57.
P163. Boulesteix M. and Biémont C. (2005) Transposable elements in mosquitoes. Cytogenet. Genome Res. 110: 500-509.
P164. Boulesteix M., Weiss M., and Biémont C. (2005) Differences in Genome Size Between Closely Related Species: The Drosophila melanogaster Species Subgroup. Mol. Biol. Evol. (sous presse).
P165. Boulétreau M. and Fleury F. (2005) Les insectes parasitoïdes et leurs partenaires multiples : des surdoués du parasitisme ? Bull. Soc. Zool. Fr 130: 177-192.
P166. Cadet P., Masse D., and Thioulouse J. (2005) Relationships between plant-parasitic nematode community, fallow duration and soil factors in the Sudano-Sahelian area of Senegal. Agriculture, Ecosystems and Environment 102: 302-317.
P167. Calteau A., Gouy M., and Perrière G. (2005) Horizontal transfer of two operons coding for hydrogenases between bacteria and archaea. J. Mol. Evol. 60: 557-565.
P168. Carvalho A., Freitas A.T., Oliveira A.L., and Sagot M.-F. (2005) A highly scalable algorithm for the extraction of cis-regulatory regions. In Proceedings of 3rd Asia-Pacific Bioinformatics Conference (APBC’05), pp. 273-282. Imperial College Press.
P169. Carvalho A., Freitas A.T., Oliveira A.L., and Sagot M.-F. (2005) An Efficient Algorithm for the Identification of Structured Motifs in DNA Promoter Sequences IEEE/ACM Transactions on Computational Biology and Bioinformatics (in press).
P170. Charif D., Thioulouse J., Lobry J.R., and Perrière G. (2005) Online synonymous codon usage analyses with the ade4 and seqinR packages. Bioinformatics 21: 545-547.
P171. Charlesworth D., Charlesworth B., and Marais G. (2005) Steps in the evolution of heteromorphic sex chromosomes. Heredity 95: 118-128.
P172. Crochemore M. and Sagot M.-F. (2005) Motifs in sequences: localization and extraction. In Compact Handbook of Computational Biology (ed. Konopka A.), pp. 47-98. Marcel Dekker Inc.
P173. Culhane A.C., Thioulouse J., Perrière G., and Higgins D.G. (2005) MADE4 : an R package for multivariate analysis of gene expression data. Bioinformatics 21: 2789-2790.
P174. David J.R., Gibert P., Legout H., Petavy G., Capy P., and Moreteau B. (2005) Isofemale lines in Drosophila: an empirical approach to quantitative trait analysis in natural populations. Heredity 94: 3-12.
P175. Décelière G., Charles S., and Biémont C. (2005) The Dynamics of Transposable Elements in Structured Populations. Genetics 169: 467-474.
P176. Dedeine F., Boulétreau M., and Vavre F. (2005) Wolbachia requirement for oogenesis : occurrence within the genus Asobara (Hymenoptera, Braconidae) and evidence for intraspecific variation in A. tabida. Heredity.
P177. Delpuech J.M., Bardon C., and Boulétreau M. (2005) Increase of the Behavioral Response to Kairomones by the Parasitoid Wasp Leptopilina heterotoma Surviving Insecticides. Arch. Environ. Contam. Toxicol. 49: 186-191.
P178. Dufayard J.F., Duret L., Penel S., Gouy M., Rechenmann F., and Perrière G. (2005) Tree pattern matching in phylogenetic trees : automatic search for orthologs or paralogs in homologous gene sequence databases. Bioinformatics 21: 2596-2603.
P179. Duponnois R., Colombet A., Hien V., and Thioulouse J. (2005) The mycorrhizal fungus Glomus intraradices and rock phosphate amendment influence plant growth and microbial activity in the rhizosphere of Acacia holosericea. Soil Biology and Biochemistry 37: 1460-1468.
P180. Duponnois R., Paugy M., Thioulouse J., Masse D., and Lepage M. (2005) Functional diversity of soil microbial community, rock phosphate dissolution and growth of Acacia seyal as influenced by grass-, litter- and soil-feeding termite nest structure amendments. Geoderma 124: 349-361.
P181. Duret L. (2005) Les trésors cachés du génome humain. Pour la Science Janvier: 16-21.
P182. Gandon S., Rivero A., and Varaldi J. (2005) Superparasitism evolution: adaptation or manipulation ? The American Naturalist in press.
P183. Ginolhac A., Jarrin C., Robe P., Perrière G., Vogel T.M., Simonet P., and Nalin R. (2005) Type I polyketide synthases may have evolved through horizontal gene transfer. J. Mol. Evol. 60: 716-725.
P184. Guéguen L. (2005) Sarment : Python modules for HMM analysis and partitioning of sequences. Bioinformatics in press.
P185. Huguet V., Gouy M., Normand P., Zimpfer J.F., and Fernandez M.P. (2005) Molecular phylogeny of Myricaceae : a reexamination of host-symbiont specificity. Mol. Phylogenet. Evol. 34: 557-568.
P186. Iliopoulos C.S., McHugh J., Peterlongo P., Pisanti N., Rytter W., and Sagot M.-F. (2005) A first approach to finding common motifs with gaps International Journal of Foundations of Computer Science (in press).
P187. Kersey P., Bower L., Morris L., Horne A., Petryszak R., Kanz C., Kanapin A., Das U., Michoud K., Phan I., Gattiker A., Kulikova T., Faruque N., Duggan K., McLaren P., Reimholz B., Duret L., Penel S., Reuter I., and Apweiler R. (2005) Integr8 and Genome Reviews : integrated views of complete genomes and proteomes. Nucleic Acids Res. 33 Database Issue: 297-302.
P188. Khelifi A., Duret L., and Mouchiroud D. (2005) HOPPSIGEN : a database of human and mouse processed pseudogenes. Nucleic Acids Res. 33 Database Issue: 59-66.
P189. Lacroix V., Fernandes C.G., and Sagot M.-F. (2005) Reaction motifs in metabolic networks. In WABI’05.
*P190. Lerat E., Daubin V., Ochman H., and Moran N.A. (2005) Evolutionary origins of genomic repertoires in bacteria. PLoS Biol. 3: e130.
P191. Lett C., Auger P., and Fleury F. (2005) Effet of asymetric dispersal and environmental gradients on the stability of host-parasitoid systems. Oikos 109: 603-613.
P192. Lopes C., Péry A.R.R., Chaumot A., and Charles S. (2005) Ecotoxicology and Population Dynamics : on the use of DEBtox models in a Leslie modeling approach Ecological Modelling 188 : 30-40.
*P193. Marais G., Nouvellet P., Keightley P.D., and Charlesworth B. (2005) Intron size and exon evolution in Drosophila. Genetics 170: 481-485.
P194. Mavingui P., Tran Van V., Labeyrie E., Rancès E., Vavre F., and Simonet P. (2005) An efficient procedure for purification of the obligate intracellular Wolbachia pipientis and representative amplification of its genome by multiple displacement amplification. Applied and Environmental Microbiology in press.
P195. Meunier J., Khelifi A., Navratil V., and Duret L. (2005) Homology-dependent methylation in primate repetitive DNA. Proc. Natl. Acad. Sci. U. S A 102: 5471-5476.
P196. Mouton L., Henri H., Boulétreau M., and Vavre F. (2005) Multiple infections and diversity of cytoplasmic incompatibility in a haplodiploid species. Heredity 94: 187-192.
P197. Mouton L., Henri H., Boulétreau M., and Vavre F. (2005) Effect of temperature on Wolbachia density and impact on cytoplasmic incompatibility. Parasitology in press.
P198. Mugnier N., Biémont C., and Vieira C. (2005) New regulatory regions of Drosophila 412 retrotransposable element generated by recombination. Mol. Biol. Evol. 22: 747-757.
P199. Nardon C., Déceliere G., Loevenbruck C., Weiss M., Vieira C., and Biémont C. (2005) Is genome size influenced by colonization of new environments in dipteran species? Mol. Ecol. 14: 869-878.
P200. Neel BD, Aouacheria A, Nouvion AL, Ronot X, Gillet G. (2005) Distinct protease pathways control cell shape and apoptosis in v-src-transformed quail neuroretina cells. Exp Cell Res.15;311(1):106-116.
P201. Nicolas M., Marais G., Hykelova V., Janousek B., Laporte V., Vyskot B., Mouchiroud D., Negrutiu I., Charlesworth D., and Moneger F. (2005) A gradual process of recombination restriction in the evolutionary history of the sex chromosomes in dioecious plants. PLoS Biol. 3: e4.
P202. Ochman H., Daubin V., and Lerat E. (2005) A bunch of fun-guys: the whole-genome view of yeast evolution. Trends Genet. 21: 1-3.
P203. Ochman H., Lerat E., and Daubin V. (2005) Examining bacterial species under the specter of gene transfer and exchange. Proc. Natl. Acad. Sci. U. S A 102 Suppl 1: 6595-6599.
P204. Peterlongo P., Pisanti N., Boyer F., and Sagot M.-F. (2005) Lossless Filter for Finding Long Multiple Approximate Repetitions Using a New Data Structure, the Bi-Factor Array. In SPIRE’05.
P205. Pisanti N., Crochemore M., Grossi R., and Sagot M.-F. (2005) Bases of motifs for generating repeated patterns with don’t cares. IEEE/ACM Transactions on Computational Biology and Bioinformatics 2: 40-50.
P206. Pisanti N., Crochemore M., Grossi R., and Sagot M.-F. (2005) A comparative study of bases for motif inference. King’s College Publications.
P207. Pisanti N. and Sagot M.-F. (2005) Network expression inference. In Applications of Combinatorics on Words (eds. Berstel J. and Perrin D.), pp. 227-250. Cambridge University Press.
P208. Sagot M.-F. and Tannier E. (2005) Perfect sorting by reversals. In COCOON’05.
P209. Semon M., Mouchiroud D., and Duret L. (2005) Relationship between gene expression and GC-content in mammals : statistical significance and biological relevance. Hum. Mol. Genet. 14: 421-427.
P210. Tannier E., Bergeron A., and Sagot M.-F. (2005) Advances on sorting by reversals Discrete Applied Mathematics (in press).
P211. Varaldi J., Boulétreau M., and Fleury F. (2005) Cost induced by viral particles manipulating superparasitism behaviour in the parasitoid Leptopilina boulardi. Parasitology 131: 161-168.
P212. Varaldi J., Fouillet P., Boulétreau M., and Fleury F. (2005) Superparasitism acceptance and patch-leaving mechanisms in parasitoids : A comparison between two sympatric wasps. Animal Behaviour 69: 1227-1234.
P213. Wertheim B., Allemand R., Vet L.E.M., and Dicke M. (2005) Effects of aggregation pheromone on individual behaviour and food web interactions : a field study on Drosophila. Ecol Entomol. in press.
P214. Zagordi O. and Lobry J.R. (2005) Forcing reversibility in the no-strand-bias substitution model allows for the theoretical and practical identifiability of its 5 parameters from pairwise DNA sequence comparisons. Gene 347: 175-182.