Sommaire de la page
Directeurs de l'axe J. Lobry, D. Chessel
Depuis sa création, le laboratoire s'est situé à l’interface entre mathématiques et informatique d’une part et biologie d’autre part. Dans ce champ multidisciplinaire, la caractéristique principale des recherches du laboratoire a été, et est toujours, l’importance attachée au pilotage de la démarche par l’objectif biologique. Ce sont avant tout des résultats biologiques que nous cherchons à obtenir. Ceci étant nous cherchons, bien entendu, à obtenir des résultats méthodologiques de portée plus générale que le cadre biologique strict qui en a motivé le développement.
Les relations entre disciplines au sein d’un projet multidisciplinaire sont complexes, les synergies difficiles à obtenir, les équilibres fragiles. Ces difficultés se traduisent par une recherche de la structuration la plus efficace qui doit rester active et sans dogmatisme. Si le contrat précédent avait cherché à regrouper les forces méthodologiques au sein d’un département, la proposition actuelle traduit l’immersion nécessaire de la méthodologie au sein même d’une grande problématique biologique. Ainsi les biométriciens (je reste attaché à ce terme en attente d’un nouveau mot dans lequel puissent se reconnaître les biomathématiciens, les biostatisticiens, les bioinformaticiens et quelques inclassables) du laboratoire sont dispersés dans chacun des trois départements proposés. Cela ne nie pas la nécessité d’une interaction forte par-delà les limites départementales, et c’est la motivation de la création de ce département transversal (j’espère que la direction du CNRS appréciera la reprise à notre modeste échelle d’un concept phare de sa dernière réforme) méthodologique.
Malgré les redondances que cela entraîne par rapport aux bilans de chacun des départements, il nous a semblé utile de faire ici un bilan (court) des activités méthodologiques du dernier contrat. Le point de vue est en effet différent ici et nous chercherons en particulier à montrer les synergies spécifiques entre activités méthodologiques et biologiques, synergies qui dépassent le plus souvent le cadre strict des équipes. Nous chercherons aussi à montrer la synergie entre des développements méthodologiques liés à des champs biologiques différents, finalement ce chapitre est une contribution à l’existence de l’interface entre informatique, mathématique et biologie en tant que champ scientifique, voire discipline. On y reconnaîtra aussi qu’étant un des deux axes constitutifs du laboratoire avec la biologie évolutive, la biométrie est un puissant moteur d’élargissement du champ d’intervention du laboratoire (certains diront de dispersion). Si nous revendiquons cette caractéristique, nous reconnaissons bien sûr qu’elle peut mériter discussion !
Cependant une description globale de la biométrie du laboratoire reste peu concrète. Nous l’avons complété ici par la description précise d’un champ particulier (l’écologie statistique) qui est bien représentative de notre point de vue sur la biométrie. Cette présentation qui constitue un hors-texte à la fin de la description de l’axe transversal, est centrée sur la biométrie à l’intérieur d’un département, bien entendu une présentation similaire aurait pu être fait pour chacun des deux autres départements.
Enfin l’axe « Méthodologie » est structurellement impliqué pour partie dans un projet INRIA : HELIX dirigé par A. Viari. Cette implication a des conséquences importantes pour le laboratoire bien entendu financières (les équipes impliquées reçoive un financement) mais surtout scientifiques par des relations privilégiées avec la partie Grenobloise d’HELIX (modélisation des réseaux de gènes, représentation des connaissances, ...) et par une association formelle d’une part avec le SIB (Swiss Institute of Bioinformatics) et d’autre part avec l’université de São Paulo (algorithmique, théorie des graphes).
Bilan méthodologique 2002-2005
Le dernier contrat a vu le renforcement de l’algorithmique avec la formalisation de l’équipe de Marie France Sagot et la nomination CR INRIA d’Éric Tannier. Le départ récent de Pierre Auger a été compensé, dans l’axe méthodologique, par la nomination MCU de Manuela Royer. Ainsi la méthodologie s’est légèrement renforcée numériquement dans ce contrat au niveau des enseignants chercheurs et chercheurs. Il faut aussi reconnaître que ce bilan est très schématique, de nombreux collègues du laboratoire étant difficile à classer dans une dichotomie (biologiste, biométricien). Au niveau du cadre technique, dans le domaine méthodologique les départs à la retraite ont été compensés (voir bilan global du labo) et un nouveau poste ITA a été créé (Stéphane Delmotte, AI). Dans la suite de la description de l’axe transversal méthodologie et pour souligner leur rôle dans la multidisciplinarité, les exemples cités seront très majoritairement choisi parmi les travaux de thèses soient récents soient en cours.
Informatique
Les premières relations avec l’informatique concernent la modélisation. L’informatique fournit des langages qui permettent de décrire la structure et le fonctionnement des systèmes biologiques. La frontière avec les mathématiques est d’ailleurs parfois un peu floue, au moins pour les biologistes que nous sommes. Par ailleurs l’informatique offre des capacités d’agir en traitant les données, en accédant efficacement aux données et aux connaissances et en réalisant des calculs massifs. Elle permet aussi une diffusion incomparable des résultats, des données et des méthodes.
Relations entre informatique théorique et biologie
L’histoire récente de ces relations au sein du laboratoire est liée au projet HELIX. Ce projet INRIA est né en 2001 et regroupe les équipes « Bioinormatique et Génomique Évolutives » et « Baobab » du laboratoire avec un groupe de l’Unité INRIA Rhône-Alpes à Grenoble. Le projet est actuellement dirigé par A. Viari (INRIA Grenoble). Le rapport d’activité d’HELIX est disponible à l’adresse d'HELIX.
L’ancrage le plus ancien dans l’informatique théorique s’organise autour de la représentation des connaissances au sein d’une collaboration avec F. Rechenmann. principalement centrée sur la cartographie génomique comparée. Cette collaboration a conduit à la soutenance de trois thèses (B. Spataro, 2001 ; G. Bronner, 2002 ; V. Navratil, 2005) et à la diffusion d’une base de données : Gem. Il faut souligner que ce travail a conduit au développement par une autre équipe du laboratoire, en collaboration avec HELIX, d’une base de données originale concernant les éléments transposables : Gate (équipe « Génomique des populations » dir. C. Biémont).
Plus récemment l’arrivée de deux chercheur INRIA et la création par M.F. Sagot de l’équipe « Baobab » a conduit à une activité forte en algorithmique (en particulier combinatoire). Les thèmes majeurs sont l’évolution des réseaux métaboliques (thèse de V. Lacroix), les mécanismes de réarrangement génomiques (thèse de C. Lemaître) et la recherche de signaux fonctionnellement signifiant dans les séquences génomique. Ce nouveau thème s’inscrit donc à la fois dans le cadre méthodologique du laboratoire mais aussi dans ses préoccupations évolutives.
Pour terminer cette description des principales relations avec l’informatique théorique nous mettons en avant le travail réalisé dans le cadre de la phylogénie moléculaire. Deux problèmes difficiles sont la maintenance incrémentale de très grands alignements comme ceux des RNAr (thèse en cours de A.M. Arigon) et la gestion de familles d’arbres. Le développement des bases d’arbres phylogéniques de familles de gènes à conduit au développement (thèse de J.M. Dufayard) d’outils de requêtes dans des ensembles d’arbres (plusieurs dizaines de milliers). Ces outils permettent entre autres une approche phylogénique de la détermination de la nature des relations d’homologie (ortho- ou para-logie).
Calcul intensif
Dans le domaine de la génomique, il est devenu banal de remarquer que la taille croissante des données et la complexité des traitements engendraient un fort besoin en calcul. De manière plus générale la volonté de prendre en compte de manière moins schématique la complexité des systèmes biologiques conduit à mettre en œuvre des méthodes d’analyse très gourmandes en ressource de calcul, en particulier de type simulation.
La politique du laboratoire dans ce domaine est claire : nous n’avons pas acquis de moyens de calculs intensifs propres (fermes ou cluster de PC) mais cherchons à utiliser les moyens de calculs existants. Cette démarche a été largement favorisée par la remarquable politique d’ouverture vers la biologie du Centre de Calcul de l’IN2P3. Le point le plus important est l’existence d’un poste d’ingénieur de l’IN2P3 dédié aux relations avec la biologie. Dans le cadre d’une convention avec le CCIN2P3, nous sommes actuellement le 11ème plus gros utilisateur du centre, ce qui vue la puissance disponible correspond à la puissance de 50 PC utilisés 24h/24, 7j/7 en continu par le laboratoire. Par ailleurs le CCIN2P3 nous assure la sauvegarde régulière des disques au travers du réseau local de l’université. Ces ressources sont gratuitement mise à disposition du laboratoire dans le cadre du soutien des centres nationaux de calculs par le département du Vivant.
Les utilisations du calcul intensif au laboratoire sont décrites au sein du bilan du département GGE. On se contentera ici de citer :
- Les calculs en amont des bases de données, en particulier ceux impliqués dans les reconstruction phylogénétiques ou des données d’expression.
- L’analyse par HMM des génomes complets de vertébrés
- Des simulations en génétique des populations ou en évolution des génomes
- Analyse statistique des structures de génomes complets (seqinR)
Diffusion et usage propre
Le principe dans la diffusion des logiciels et des bases de données par le LBBE a toujours suivi la même voie : l’objet informatique est créé pour répondre à un besoin de recherche interne puis amélioré et étendu pour pouvoir être diffusé. Cette stratégie, que d’aucuns trouveront artisanale, a ses avantages. Le principal est sans aucun doute la pérennité des services rendus à la communauté. Ainsi ACNUC est une base de données de séquences génomiques née dans les années 70 et toujours largement utilisée, ADE4 logiciel statistique dédié à l’analyse de données date de 1985 et continue d’évoluer aussi bien dans son contenu que dans l’efficacité de sa diffusion. Le deuxième avantage, beaucoup plus discutable, est que cette stratégie ne coûte pratiquement rien aux tutelles et peut être mis en œuvre par un laboratoire sous-doté en ingénieurs (AI+IE+IR (ITA+IATOS) = 5).
Dans ce contexte le recours à une plateforme de bioinformatique est une solution naturelle. Le PRABI (Pole Rhônes-Alpes de Bioinformatique) est une plateforme opérationnelle RIO qui s’appuie sur 12 équipes de recherche de la région. Cette plateforme a été dotée de locaux sur le site de Gerland et bientôt sur le site de la Doua, par contre son potentiel en personnel est très faible : un seul poste IR prévu en fin 2006 et dédié à la pharmaco-génomique. Ainsi l’aide de cette plateforme reste faible et, actuellement ce sont plus les moyens des laboratoires qui font vivre la plateforme que le contraire.
Avant de donner un tableau des principales productions informatiques du laboratoire, sont présentés les principaux modes de diffusion utilisés.
accès par Web :
Des interfaces permettent au travers d’un navigateur standard de réaliser des interrogations de bases de données ou des calculs sur le serveur du PBIL/Doua. Un espace disque temporairement alloué au « visiteur » lui permet d’enchaîner plusieurs tâches
logiciel à télécharger :
Ces logiciels doivent être utilisés localement, ce sont des bibliothèques de routines (Sarment en Python), ou des « packages » R (ADE 4, seqinR).
la vigueur hybride :
Plus récemment, s’est développée au laboratoire une démarche intégrant les deux précédentes. L’idée est de fournir des outils de développements dédiés à un domaine donnés et qui offre de manière transparente à l’utilisateur un accès aux bases de données associées et gérées par le PRABI. On peut citer 3 exemples : i) une bibliothèque C permettant l’accès à l’ensemble des bases gérées sous ACNUC ; ii) l’interfaçage avec Seqinr qui permet le développement sous R avec des requêtes sur les bases de séquences ; iii) le projet GeM qui fournit un ensemble de fonctions R permettant l’analyse statistique de données de cartographie comparée et des possibilités de requête sur la base GeMCore gérant l’ensemble des génomes complétement séquencés de vertébrés.
| Nom | nature | Dpt | Remarques |
|---|---|---|---|
| ACNUC | BD | GGE | Base de données à la base de la plupart des bases génomiques du laboratoire, diffusion Web + Client serveur |
| ADE 4 | Logiciel statistic R package | EE | « gros » logiciel implémentant les méthodes d’analyse de données dites vectorielles avec une référence particulière à l'écologie |
| ADEHabitat | Package R | EE | Sélection d’habitat |
| ADS | Package R | EE | Analyse spatiale |
| BIBI | BD + identification bactérienne | BMS | Outil d’identification bactérienne à partir de séquences, application médicale, Web |
| CpGProD | Logiciel | GGE | Identification des ilôts CpG |
| DYNATICA | Mathematica | BMS+GGE | Metalanguage de simulation de problèmes dynamiques et de sorties graphiques |
| GEM | Base de données | GGE | Cartographie génomique comparée |
| Hogenome, Hovergen, Homolens | Base de données | GGE | Familles de gènes et phylogénies |
| Hoppsigen | Base de données | GGE | Rétropseudogènes |
| Lalnview | Webiciel+local | GGE | Comparaison de séquences |
| Oriloc | Webiciel+local | GGE | Origine de réplication |
| Philowin | C | GGE | Phylogénie moléculaire |
| RAP | Java | GGE | Réconciliation d’arbres phylogéniques |
| RFIT | R | BMS+GGE | estimation des paramètres de modèles différentiels |
| Sarment | Bibliothèque Python | GGE | HMM |
| Seqinr | Package R | GGE | Analyse de séquences |
| SMILE | Logiciel C | GGE | Recherche de motifs complexes dans les séquences |
Systèmes dynamiques
La prise en compte du temps (ou de l’espace sur une ligne comme la séquence d’un génome) est, bien évidemment, une nécessité dans l’analyse de l’évolution des systèmes biologiques. On retrouve donc cette problématique dans les trois départements. Deux champs techniques, pas totalement indépendants, se séparent traditionnellement sur la prise en compte ou non de phénomènes stochastiques. De 2002 à septembre 2005 la modélisation déterministe au laboratoire a été principalement animée par P. Auger qui a depuis pris la direction d’un laboratoire de l’IRD. C. Lett, MCU, spécialiste des systèmes multi-agents est détaché à l’IRD depuis 2003. Actuellement, même si les systèmes déterministes sont largement utilisés au sein du laboratoire, les développements méthodologiques reposent principalement sur S. Charles (MCU) qui a rejoint l’équipe Baobab. Même si le développement méthodologique reste actif grâce au travail de S. Charles le domaine est fragilisé dans le cadre du développement d’approches mathématiques nouvelles en biologie. L’effort de développement méthodologique porte en effet plus actuellement au laboratoire sur les modèles stochastiques. Dans le cadre de l’utilisation d’outils déjà bien implanté en biologie, il faut remarquer un effort pour concilier le besoin de représentation de la complexité biologique et la necessité de modèles relativement simples permettant des interprétations biologiques qui aillent au delà d’une simple constatation d’adaptation du modèle aux données. Des exemples de ce souci pourront être trouvés dans chacun des départements, on peut par exemple renvoyer à l’utilisation de modèlles d’EDO avec retard pour la modélisation des relations hôtes parasites (EE)
D’une manière assez générale, une caractéristique majeure des développements du domaine est l’hybridisme entre des outils différents. Par exemple des modèles en temps continu (équations différentielles ordinaires) vont modéliser les effets de polluants sur les traits d’histoires de vie de daphnies, les paramètres démographiques seront ensuite intégrés à des modèles de type Leslie couplant ainsi effet des polluant et la dynamique de population (thèse en cours de E. Billoir).
Dans le domaine de la santé, la nécessité de modéliser simultanément des phénomènes de natures différentes conduit à des modèles probabilistes complexes. Un des facteurs majeurs de cette complexité vient de la structure même des observations. Ainsi dans la modélisation d’une infection verticale par le sida (thèse en cours de M. Tournoud), la modélisation de l’infection elle même doit prendre en compte de multiples facteurs (en particulier la possibilité de contact multiple avec le virus au cours de l’allaitement) mais le modèle doit également intégrer la connaissance partielle de la date de détection du virus due aux intervalles de temps entre les prises de sang.
L’aspect stochastique du processus devient central quand la variabilité des différentes trajectoires est l’objet même de l’étude. En épidémiologie, l’étude de l’extinction du parasite en population finie d’hôtes est un exemple que l’on retrouve dans le département « Écologie Évolutive » mais qui pourrait aussi renvoyer à des domaines relevant de la santé humaine. Dans le cas de virus de chats (thèse de F. Sauvage), des chaînes de Markov en temps continu ont été étudiées, principalement par simulation, pour modéliser l’évolution de populations constituées d’individus sensibles ou infectés.
Les limitations des chaînes de Markov dans la représentation de situations complexes a conduit l’utilisation de réseaux de Pétri, et des HMM.
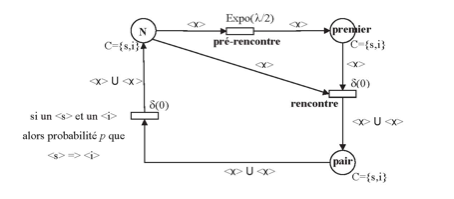
Les réseaux de Pétri stochastiques colorés (thèse de N. Bahi-Jaber) ont permis de modéliser des situations en épidémiologie « écologique » avec un nombre important de statuts d’individus. L’aspect graphique du modèle permet de plus une compréhension simple du modèle par des biologistes non-spécialistes. Un exemple de ce type de modèle est montré dans la figure suivante dans le cas des relations entre sensibles (s) et infectés (i).
Les HMM (Hidden Markov Model) sont largement utilisés pour modéliser des systèmes biologiques, en particulier dans le cadre du génome. Leur représentation graphique (finalement assez proche des « modèles à compartiments »), leur capacité à représenter des hétérogénéités et l’existence de quelques algorithmes efficaces en font actuellement l’une des meilleures approches pour estimer la nature de régions génomiques (prédiction de gènes par exemple). La démarche actuelle au sein du laboratoire consiste à utiliser ces modèles plus comme outils d’analyse que de prédiction. L’objectif est moins de structurer les génomes en régions statistiquement homogènes que de comprendre les mécanismes de cette structuration. Ce travail a donné lieu au développement d’une bibliothèque de fonctions librement disponible (Sarment) et une thèse (C. Melo de Lima, 2005).
Modélisation statistique
Les statistiques apparaissent comme le lien entre modèles et théories d’une part et données d’autre part. En biologie, les situations dans lesquelles la variabilité « non contrôlée » n’existe pas sont rares. Les variabilités individuelles, expérimentales ou encore de mesures nécessitent dans leur prise en compte un raisonnement statistique. Au-delà de la présentation « traditionnelle » d’aide à la décision (je rejette ou j’accepte), la statistique joue un rôle majeur dans les démarches de sélection de modèles, de résumé et d’exploration de données, et est le plus souvent complètement intégrée aux modèles probabilistes.
L’activité statistique est l’activité méthodologique la plus ancienne dans le laboratoire. Il a semblé que l’explicitation de la démarche méthodologique du laboratoire serait éclairée par la description plus complète d’un champ particulier, l’écologie statistique a été choisie. Ainsi un long encart est présenté à la fin de la présentation du département, son objectif est, au travers d’une activité méthodologique majeure, de discuter plus en détail des questions transversales à l’activité du laboratoire. Les autres aspects des statistiques du laboratoire seront présentés brièvement maintenant.
L’interaction très forte entre utilisateurs et biométriciens dans le domaine de la santé crée des besoins méthodologiques particuliers, et donc réponses originales décrites dans le chapitre sur le département BioMathSanté. En particulier la difficulté de maîtriser les plans expérimentaux, la multiplicité des approches sur un même objet conduisent à des concepts de méta analyse et indirectement à une utilisation forte des modèles bayésiens. On peut remarquer que les mêmes contraintes commencent à apparaître en écologie, qui devient également forment demandeuse d’approches bayésiennes, même si dans de nombreux cas le besoin est plus technique et ne résulte pas d’une réelle volonté de modéliser des connaissances a priori.
La statistique intervient naturellement comme complément d’une modélisation utilisant d’autres formalismes soit dans la phase d’estimation soit dans la validation. Ainsi le laboratoire utilise la modélisation par modèle de Markov aussi bien dans l’estimation des phylogénies que dans la segmentation des génomes. Dans le premier cas, l’objectif est de mieux prendre en compte les connaissances sur les processus évolutifs du génome dans la reconstruction de l’histoire des espèces. En particulier les variations au cours du temps des fréquences des bases nucléotidiques conduisent à l’utilisation des chaînes de Markov non homogènes et non réversibles. Le problème en cours est de développer, à partir de méthodes au maximum de vraisemblance efficace dans le cas réversible (PHYML, Guindon, Gascuel 2003), des méthodes qui s’affranchissent de cette contrainte. Dans le cadre de la segmentation des génomes le problème posé était plus simple, c’est un problème de choix de modèles chacun d’eux étant un HMM « caractéristique » d’une classe d’isochore. La méthode utilisée a été une méthode bayésienne « simple ».
Perspectives de l’axe
Fonctionnement de l’axe transversal
L’objectif est de favoriser le partage de l’expérience méthodologique au sein du laboratoire entre les trois départements, en particulier de gérer la difficulté de l’éloignement des sites du laboratoire Bio-Math-Santé du campus de la Doua. On espère ainsi :
- Favoriser le transfert de méthodes pratiquées ou développées d’un domaine d’application à une autre.
- Favoriser l’émergence de nouvelles méthodes par confrontation d’idées.
- Favoriser une meilleure intégration entre les aspects recherche, enseignement et formation continue.
- Favoriser une démarche qualité dans le domaine de l’analyse des données et de la production de logiciels.
Au-delà d’une démarche « classique » de réunion de travail et de séminaires, l’enseignement d’une part et les codirections de thèses d’autre part sont deux puissants moyens d’interaction entre équipes méthodologiques. Le développement de matériel pédagogique adapté à la biométrie (fiches, site web, …) permet un premier transfert qui ouvre ensuite à la discussion pointue. Il faut enfin souligner que la disponibilité de la nouvelle salle de formation du PRABI permettra la mise en place beaucoup plus simple d’une formation continue en méthodologie.
Perspectives scientifiques
Il n’est pas question ici de reprendre les axes méthodologiques présentés dans chacun des départements. On peut cependant mettre l’accent sur quelques champs méthodologiques que nous comptons explorer dans les quatre ans à venir.