Here are the main themes of my research :
OriSeq project
Functional Data analysis
CGH microarrays analysis and segmentation models
Biological networks, random graphs and motifs
OriSeq Project
In a recent project we analyze Oriseq data, and we aim at determining epigenetic signatures that characterize the spatio temporal program of replication. Tables of our analysis can be found here : K562 table (.bed), IMR90 table (.bed), Hela table (.bed), CGI table (.bed), as well as their description. Our method of peak detection using scan statistics can be reproduced using this code (with a toy example). The comparison of our method with the results of SoleSearch can be found here.
Functional Data analysis
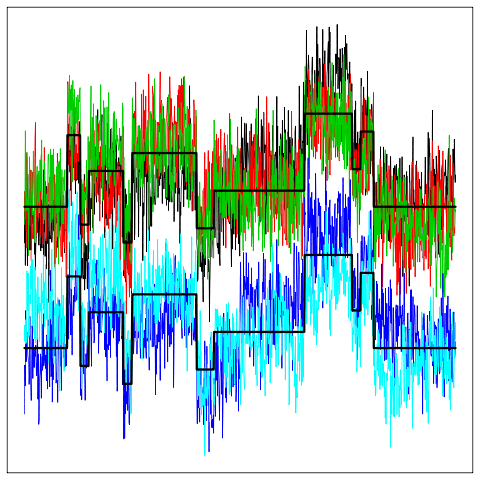
| We are currently studying curve clustering models that account for an inter-individual variability throught mixed functional models.
In this setting the aim is to cluster data which are in the form of curves and we introduce a random effect term to model the inter-individual variability.
Traditional random effects used in mixed linear models here become random processes. We use a wavelet-based representation of this model. The challenge
is to propose a reliable clustering procedure for this type of data and to develop accurate dimension reduction techniques to overcome the high dimensionality
of the model. We are also developing an R package dedicated to curve clustering.
Collaborators on the subject: Sophie Lambert-Lacroix (LJK-IMAG), Guillemette Marot (PostDoc LBBE), Madison Giacofci (PhD Student, LJK-IMAG). |
 |
CGH microarrays analysis and segmentation models
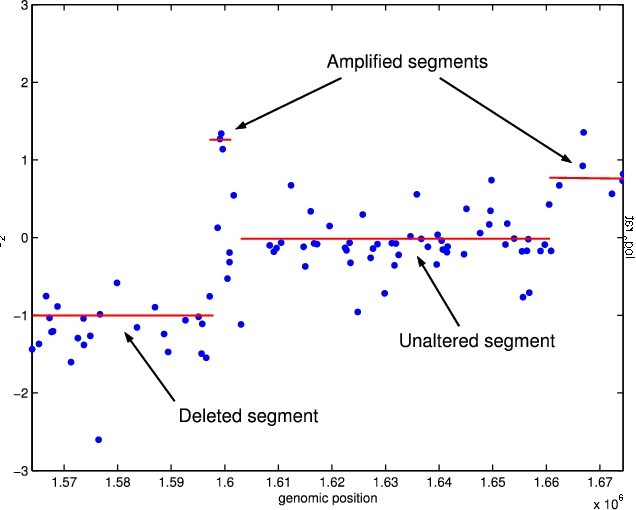
| The CGH microarray technology aims at mapping chromosomal aberrations at the genome level.
When dealing with one sample analysis, the purpose is to detect the aberrations, and to
give their boundaries as precisely as possible. Another challenge is to assess the
biological status of the detected segment (deleted/normal/gained/amplified). Then the
purpose can be to analyse multiple sample datasets. Actually, this consitutes an important
main motivation of array CGH experiments, that is the association between chromosomal
aberrations and clinical outcome. An introduction to array CGH data analysis can be found here
Segmentation models have been very successful in the analysis of array CGH data. They are used to map abrupt-changes along a signal at unknown locations on the genome. Theoretical questions associated with segmentation models are : finding an efficient algorithm to find the breaks, and estimating the number of breaks. For the first aspect, we have been interested in the use of Dynamic Programming, and in model selection for the second aspect. An introduction on segmentation models can be found here Collaborators on the subject: Stephane Robin (AgroParisTech) Emilie Lebarbier (AgroParisTech), Mark Hoebeke (INRA). |
 |
Biological networks, random graphs and motifs
|
With the increasing power of high throughput technologies and storage capacities, it is now
possible to explore datasets which are in the form of complex networks. Many scientific fields
are concerned by these major advances, such as physics, social sciences, and molecular biology.
One characteristics of interest when studying complex networks is their topology or the way
particules, proteins or social agents interact. More generally, studying the topology is
crucial to understand the organization of networks, as structure often affects function.
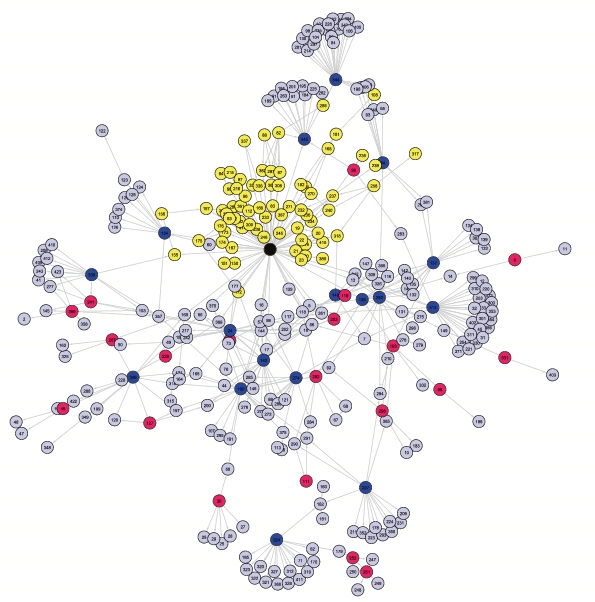
Random graphs constitute a tool of choice for this purpose and we developed a particular random
graph model that is called MixNet. The aim of MixNet is to cluster nodes of a network that share the same connectivity profile is done via a generalization of mixture models to network data. As a result, MixNet provides a synthetic picture of the main connectivity patterns that make the network. A challenge when using these models lies in the optimization algorithm to estimate the parameters. I worked on a variational version of the EM algorithm for this purpose, and on online strategies to speed up its execution. The applications are diverse, from molecular networks (Transcription Regulatory Networks) to foodwebs and weblogs. Another way to study the architecture of networks is to study their basic subunits, building blocks or network motifs. The statistical question associated with network motifs is the question of their exceptionality. Are there motifs that occur more than expected in biological network ? This question is related to the distribution of the count of some subgraphs in random graphs. Collaborators on the subject: Stephane Robin (AgroParisTech) Jean-Jacques Daudin (AgroParisTech), Vincent Miele (LBBE), Christophe Ambroise (Evry), and more generally the SSB group. |
 |